Introduction
Wilson's disease (WD) is a rare genetic disorder caused by a pathogenic mutation of the ATPase copper transporting beta (ATP7B) gene, which is involved in cellular copper metabolism. Because of this mutation, a defective protein is synthesized, which does not allow the incorporation of copper ions into ceruloplasmin, as well as the excretion of the metal through bile. Thus, the abnormal copper metabolism subsequently leads to the accumulative deposition of copper in the target organs and impairs the normal functions of the affected organs, especially in the liver and brain [1].
The prevalence of WD in the general population is 1:30 000 - 200 000 people [2, 3], but it can vary according to the geographical area, and in socially, economically, culturally, religiously, and geographically isolated communities it can be much lower, especially if marriages between relatives are practiced [4].
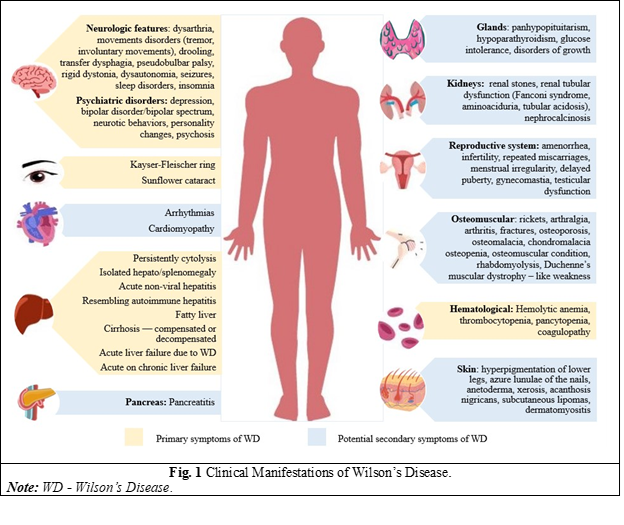
A polymorphic clinical picture characterizes WD, and the disease can evolve from asymptomatic forms, isolated non-specific symptoms, to acute liver failure. It must be suspected in all adults or children presenting with unexplained liver disease or/with a movement disorder of uncertain cause, neuropsychiatric disorders, or unexplained hemolytic anemia [5]. Because of its broad spectrum of clinical manifestations that can present in almost any decade of life, a high degree of clinical suspicion is needed for diagnosis (Fig. 1) [6, 7]. Despite some specific changes in investigations, the diagnosis of WD remains a challenge, given that there is no single specific test, but several tests are needed, and genetic testing for ATP7B mutations is an important criterion when routine examinations are not well defined [8].
In 1912, British neurologist Samuel Kinnear Wilson first described WD as “progressive lenticular degeneration”, a fatal familial neurological disorder that may be associated with chronic liver disease leading to cirrhosis. In his work he mentioned that “the most curious and remarkable feature of this familial nervous disease is the constant presence of a profound degree of cirrhosis of the liver”, but at that time it was considered not to cause clinical problems [9]. In 1916, physician Byrom Bramwell reported a family in which four siblings died of "acute fatal cirrhosis". Between 1925 and 1929, Drs. Barnes and Hurst described a family in which 3 of the eight children had Wilson's disease, but liver lesions preceded neurological symptoms, and the fourth child died of severe liver disease without neurological symptoms [10]. Respectively, the involvement of several members of the same family with this disease is observed, which denotes the importance of screening for the whole family.
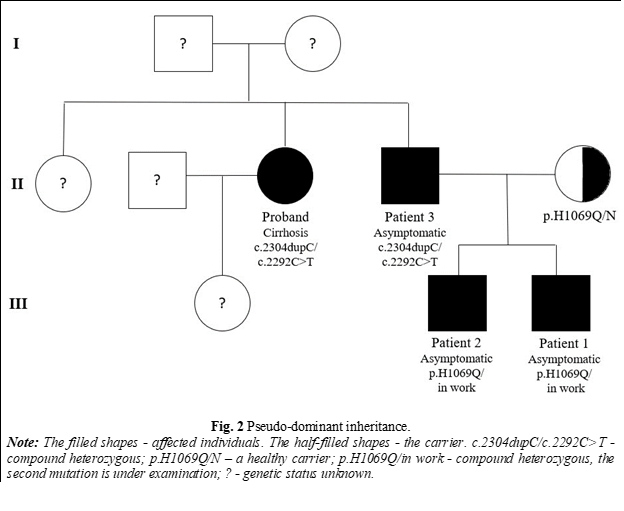
WD is an autosomal recessive disorder. Although it is a monogenic disorder, and the inheritance of characters occurs according to Mendelian laws, paradoxical transmissions of the disease, such as pseudo-dominant transmission, have been recorded. Thus, cases of WD have been reported in consecutive generations, but this can occur, particularly when carrier frequencies are as high as in WD [11]. In addition, cases of atypical inheritance have been described, such as the presence of three concurrent mutations in a single patient or segmental uniparental disomy. This change occurs when both homologs of a chromosome come from the same parent [12].
It is important to screen family members of a newly diagnosed patient with WD, because the risk of developing this disease is 25% for siblings, and 0.5% for offspring [3]. Thus, screening first-degree relatives can identify persons affected by WD in the asymptomatic phase, although organ lesions may already be present. In this phase, the evolution of the disease is favorable with the initiation of specific treatment. Therefore, it is essential to identify them as early as possible and start chelator therapy [13]. This also increases disease awareness among family members, allows for close medical surveillance, and differentiates healthy members or healthy carriers who might be potential donors if a sick member requires a liver transplant [14]. At this stage, it is important to differentiate asymptomatic patients from healthy carriers because 15% of carriers show changes in copper parameters [15]. Genetic testing can confirm the diagnosis when biochemical testing is inconclusive and can identify the individual's status as simple heterozygous, compound heterozygous, or homozygous recessive [13].
Taking into account the heredity, phenotypic diversity, and genotypic heterogeneity of the disease, our goal was to evaluate the families of patients diagnosed with WD to initiate appropriate treatment in asymptomatic members.
Materials and methods
12 families were evaluated retrospectively and prospectively, between 2008 and 2022 within the Human Molecular Genetics Laboratory and the Gastroenterology Discipline of the Nicolae Testemiţanu State University of Medicine and Pharmacy. The given study is part of the research protocol that obtained a favorable opinion from the Research Ethics Committee (minutes No.1 dated 25.05.2021). All study participants (parents of children or patients) signed informed consent forms.
Inclusion criteria:
Presence of a sick member with WD;
Patients aged ≥ 5 years;
Consent of the patient or legal representative to participate in the study.
Exclusion criteria:
Absence of family members affected by WD;
Patient aged < 5 years;
Lack of patient or legal representative consent to participate in the study.
The investigation methods used within the study were:
clinical examination – patients' complaints, medical and family history were collected. Physical examination was performed with recording of anthropometric data and vital signs;
laboratory investigations – complete blood count, liver biochemical profile, coagulation, and copper parameters. Liver biopsy with copper quantification in dry liver tissue was not performed for technical reasons, it is not available in the country.
instrumental examinations – evaluation of Kayser-Fleischer rings of the cornea, abdominal ultrasonography, Fibroscan, and brain magnetic resonance imaging.
molecular-genetic analysis –Sanger sequencing of the ATP7B gene was performed, examining exons with a high and moderate frequency of mutations, at the Human Molecular Genetics Laboratory in Moldova. In certain cases, whole gene sequencing was performed in Germany, Italy, and France.
D- Penicillamine test – The first dose (D-penicillamine 500 mg) is administered at 8:30 a.m., and the next dose (D-penicillamine 500 mg) at 8:30 p.m. (12 hours after the first dose). Urine collection begins after the first dose, in a special "acid-washed" container, so care is required in handling. Collect continuously for 24 hours, including night and morning samples.
calculation of the Leipzig Scoring System (2001) for all enrolled subjects; a score ≥ 4 points establishes the diagnosis of WD.
mathematical-statistical processing methods – Microsoft Excel, Epi Info, Student's t-test.
creation of the figures - Canva, PowerPoint.
Results
All individuals included in the study underwent clinical, paraclinical, and genetic examinations. Each patient was of Caucasian origin, and originally from Moldova. Most probands originate from the south of the country (6/12), and 3 probands each originate from the north and the center of the country. No patient reported consanguineous relationships. Hepatic onset was more common in females (4/6, p < 0.01), while neurological onset was more prevalent in males (4/6, p < 0.05).
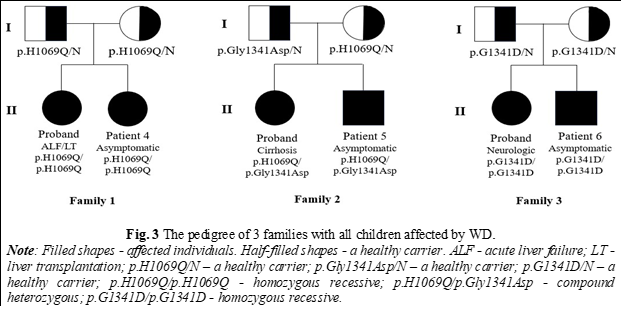
In 9 of the families, the 1st-degree relatives were tested - parents and siblings, in the other 3 cases only their descendants were evaluated. In half of the cases (6 out of 12), both parents are healthy carriers; in the other 3 families, one parent is a healthy carrier, but the other parent has not been tested. Among the siblings, 4 were identified as healthy carriers and 2 were healthy (no mutation detected). The most frequent mutations detected were p.H1069Q and p.G1341D, both as homozygous recessive and heterozygous (simple and compound). A rare mutation has been detected - c.2292C>T.
Table 1. Results of investigations of new patients with WD identified by family screening. | |||||||
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 |
Age, y | 5 | 8 | 34 | 12 | 5 | 23 | 22 |
Sex | Male | Male | Male | Female | Male | Male | Male |
Relationship with proband | Nephew | Nephew | Brother | Sister | Brother | Brother | Cousin |
Clinical symptoms | No | No | No | No | No | No | Yes |
Phenotype | Hepatic | Hepatic | Hepatic | Hepatic | Hepatic | Hepatic | Neurohepatic |
Serum ceruloplasmin | 10 (15-30mg/dl) | 14 (15-30 mg/dl) | 5 (15-30 mg/dl) | 24 (16-45 mg/dl) | 24 (16-45 mg/dl) | 6.9 (25-43 mg/dl) | 53 (200-600mg/l) |
Urinary copper 24 h | 72.7 (3-35 µg/24h) | 117.95 (10-60 µg/24h) | 139.4 (10-60 µg/24h) | 41.91 (10-60 µg/24h) | 41.91 (10-60 µg/24h) | 52.4 (10-60 µg/24h) | 189 (<60 ug/24h) |
Urinary copper 24 h after D-Penicillamine test | 424.9 (10-60 µg/24h) | 976.5 (10-60 µg/24h) | 484.9 (10-60 µg/24h) | 1022 (10-60 µg/24h) | 325 (10-60 µg/24h) | 1324 (<60 mg/24h) | 317 (15-59 µg/24h) |
Serum ALT | 185 (<36 U/L) | 173 (<29 U/L) | 38 (<41 U/L) | 100 (<30 U/L) | 20 (<30 U/L) | 58.8 (<35 U/L)) | 19.9 (<35 U/L) |
Serum AST | 88.6 (<53 U/L) | 104 (<36 U/L) | 26 (<37 U/L) | 39 (<30 U/L) | 15 (<30 U/L) | 35 (<31 U/L) | 20.2 (<35 U/L) |
Kayser-Fleischer ring | Absent | Absent | Absent | Absent | Absent | Absent | Present |
Abdominal echography | Normal | H-megaly | HS-megaly | H-megaly | HS-megaly | HS-megaly | S-megaly |
Cranial magnetic resonance imaging | Normal | Normal | Normal | Normal | Normal | Not done | diffuse cerebral atrophy in both cerebral and cerebellar hemispheres |
ATP7B genotype | p.H1069Q/ in work | p.H1069Q/ in work | c.2304dupC/ c.2292C>T | p.H1069Q/ p.H1069Q | p.H1069Q/ p.Gly1341Asp | p.G1341D / p.G1341D | p.H1069Q/ p.G1341D |
Leipzig Score | 5p | 4p | 8p | 6p | 6p | 8p | 12 p |
Note: ATP7B - ATPase Copper Transporting Beta; H-megaly – hepatomegaly; HS-megaly – hepatosplenomegaly; S-megaly – splenomegaly; ALT - alanine transaminase; AST - aspartate transaminase, y – years, h – hour. | |||||||
As a result of the evaluation in 5 out of 12 families involved in the study, 7 new members with WD were identified (6 males and 1 female). The mean age was 16 years (range 5-34 years). In 4 cases, there were 1st-degree relatives (siblings), and in 3 cases - 2nd-degree relatives (cousins, nephews). 6 patients were asymptomatic, and 1 had neurological symptoms but were not included in any nosology until the evaluation for WD (Table 1). In the case of one family, a paradoxical transmission of the disease was identified - pseudo-dominant inheritance (Fig. 2). In 3 cases where both parents are healthy carriers, both children were diagnosed with WD, with one diagnosis occurring through family screening (Figure 3). In 6 cases, WD was associated with liver damage, and one case presented with a mixed phenotype. In 2 asymptomatic patients, no changes in copper parameters were observed, only cytolysis with hepatosplenomegaly or only hepatomegaly being highlighted, but after stimulation with D-penicillamine, urinary copper in 24 hours increased more than 5 times the upper limits of the norm. In both cases, the genetic test confirmed the presence of 2 pathogenic mutations. Kayser-Fleischer ring was observed only in a single patient with neurological damage. All patients newly diagnosed with Wilson's disease initiated specific therapy.
Discussion
Genetic counseling is essential for families of patients with WD, and screening of first-degree relatives is recommended by all international guidelines on diagnosis and treatment of WD [3, 5, 13, 14, 16]. Accurate and timely diagnosis of WD is important for the affected person's relatives, as it enables the most favorable treatment outcomes. [6]. The assessment algorithm includes a comprehensive clinical and biochemical evaluation, as well as an analysis of the ATP7B genotype [16] (Fig. 4).
Newborn screening did not yield the expected results, so the running of national programs is not justified [14]. Screening should be delayed until the age of 2 years, sometimes even later, because WD is rarely symptomatic until 5 years [16]. Occasionally it can be initiated earlier, if there are obvious signs of liver or occult damage (isolated hepatomegaly or splenomegaly, or fatty liver in the absence of changes in biochemical tests) [14]. In special cases, prenatal as well as neonatal testing based on genotype analysis can be performed [5].
WD can be transmitted from affected but asymptomatic parents to their offspring. First-degree relatives do not include only the siblings of a proband, but also the offspring and parents of the proband. Although the risk of developing the disease is higher in siblings, the risk to parents (0.5%) and offspring (0.5%) is underestimated. Despite this, screening of parents and children of a proband is warranted considering the lethal potential of WD [17].
Most commonly, parents are considered healthy carriers. However, there have been reports of patients over 40 years diagnosed with WD. Therefore, considering the possibility of a late onset, clinical and paraclinical exploration of the parents of a newly diagnosed child with WD is indicated [18]. Parents of the proband should contact their siblings to inform them that they may be carriers of Wilson's disease and should be referred for family screening [5]. Also, it should be noted that for patients identified with WD in childhood or adolescence, it is recommended to test their descendants, once they decide to have children [17].
The identification in 2 consecutive generations of WD in apparently unrelated families suggests the advantage of WD screening in the offspring of an affected parent [16]. This was also observed in Patient 1 and Patient 2 in the reported study. Therefore, systemic family screening is recommended despite the autosomal recessive nature of WD transmission [8].

WD can present with different clinical symptoms and sometimes different phenotypes in patients with the same genotype, even within the same family [17, 18]. This variability was also observed in our study group, especially in families where both children were affected by WD (Fig. 2 and 3). According to the Mendelian laws of autosomal-recessive transmission, in the case of carrier parents, there is a 25% chance for offspring to develop WD, but in these families, both children were diagnosed with this disorder. This highlights the genetic complexity of the disease, as well as the involvement of potential epigenetic factors.
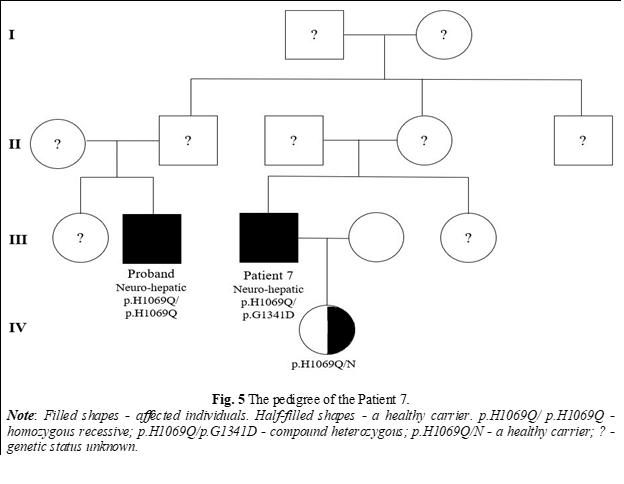
The probability of nephews and nieces being affected by WD is 1 in 600, and for cousins, the probability is 1 in 800 [19]. In 2 different families in our research with affected members, 2 nephews were identified with WD (Fig. 1) and a cousin (Fig. 5). No consanguineous marriages were reported in these families. The literature describes a case where, after family screening, new WD diagnoses emerged across generations in a single family, including an uncle and two cousins [20]. Such cases highlight the importance of testing distant relatives, especially in more isolated areas.
In our study, it was identified a rare synonymous ATP7B sequence variant c.2292C>T (p.Phe764=) in association with the pathogenic variant c.2304dupC (p.M769Hfs*26) at Patient 3. His sister (proband) has the same mutations. This variant c.2292C>T (p.Phe764=) increases the rate of exon 8 skipping in the canonical ATP7B transcript, predicting an ATP7B protein lacking transmembrane domains 3 and 4 [21]. This mutation accounts for ~0.5% (14 of 2816) of ATP7B alleles evaluated in a study of WD patients [22, 23]. The research of M. Panzer and her colleagues highlighted that the synonymous sequence ATP7B variant c.2292C>T is pathogenic by affecting messenger ribonucleic acid (mRNA) splicing. It is associated with WD being heterozygously compounded with other pathogenic variants or homozygous for this sequence variant [21].
Detection of two pathogenic or probably pathogenic variants on both chromosomes confirms the diagnosis of WD, although, in large studies of Caucasian patients with WD, the pathogenicity of both ATP7B alleles was associated in approximately 80% of patients tested [22, 23]. Previous studies reported that in 1-21% of cases, no pathogenic variant or only one ATP7B variant was detected. Thus, differentiating between WD patients and healthy carriers is a challenge in the presence of ambiguous symptoms or laboratory tests [12, 22, 23]. A plausible explanation for missing variants in Sanger sequencing of all exons and exon-intron border regions is the presence of genetic deletions/duplications that can only be detected by multiplex ligation-dependent probe amplification (MLPA) or the presence of mutations in untranslated regions, the promoter region or the deep intronic region [24].
Up to 15% of healthy carriers may show mild biochemical changes. Consequently, in such cases, it is difficult to establish the diagnosis of WD or carrier, despite a complex molecular-genetic analysis. In addition, the genetic test is not always available, or the result may be received much later [25]. In some situations, such as biochemical abnormalities and the absence of mutations or the presence of only one, it is recommended to perform a liver biopsy with copper quantification in the dry liver tissue. However, the decision to perform an invasive examination on an asymptomatic person is difficult and uncertain [13]. Thus, there is a need for an accurate, reliable, and non-invasive biological tool for family screening.
Exchangeable copper (CuEXC) is a biochemical marker that estimates free copper overload and provides data on the severity and dissemination of WD. Relative exchangeable copper (percentage of CuEXC to total serum copper) appreciates the toxic fraction of copper in blood and is an excellent biomarker for WD diagnosis with 100% specificity and 100% sensitivity [26]. Research by Dr. Trocello and colleagues showed that REC could be a biomarker that statistically significantly differentiates WD patients from simple heterozygotes (P = 0.016), as well as WD patients from healthy individuals (P = 0.015) so it can be useful for family screening. However further studies are needed to validate this test for family screening [25].
The study had limitations related to the small number of patients examined, therefore to obtain a more accurate result; it is recommended to have evaluated a large sample size. It was also not possible to clinically examine and genetically test all first-degree relatives of the patients.
Conclusions
Our study showed that family screening plays a significant role in identifying asymptomatic members with WD. Genetic testing should be performed to differentiate asymptomatic patients with WD from healthy carriers.
Abbreviations
ATP7B - ATPase Copper Transporting Beta, CuEXC - exchangeable copper, MLPA - multiplex ligation-dependent probe amplification, mRNA - messenger ribonucleic acid, WD - Wilson's disease.
Competing interests
None declared.
Patient consent
Obtained.
Ethics approval
This study was approved by the Research Ethics Committee of Nicolae Testemițanu State University of Medicine and Pharmacy (minutes no.1, from 25.05.2021).
Authors’ contribution
AT conceived the study and participated in the study design. VS conceived the study, performed the genetic tests, and participated in the study design. VC participated in the study design, performed the statistical analysis, and drafted the manuscript. All authors have read and approved the final version of the article.
Acknowledgements and funding
No external funding.
Authors’ ORCID IDs
Veronica Cumpata – https://orcid.org/0000-0003-1921-8192
Adela Turcanu – https://orcid.org/0000-0002-7684-1768
Victoria Sacara – https://orcid.org/0000-0001-9200-0494
References
Wu F, Wang J, Pu C, Qiao L, Lang C. Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015;16(13):6419-6431. doi: 10.3390/ijms16036419.
Gomes A, Dedoussis G. Geographic distribution of ATP7B mutations in Wilson disease. Ann Hum Biol. 2016;43(1):1-8. doi: 10.3109/03014460.2015.1051492.
European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J Hepatol. 2012;56(3):671-685. doi: 10.1016/j.jhep.2011.11.007.
Cocoș R, Șendroiu A, Schipor S, Bohîlțea LC, Șendroiu I, et al. Genotype-phenotype correlations in a mountain population community with a high prevalence of Wilson’s disease: genetic and clinical homogeneity. PLoS One. 2014;9(6): e98520. doi: 10.1371/journal.pone.0098520.
Shribman S, Marjot T, Sharif A, Vimalesvaran S, Aftab A, Graeme A, et al. Investigation and management of Wilson’s disease: a practical guide from the British Association for the Study of the Liver. Lancet Gastroenterol Hepatol. 2022;7(6):560-575. doi: 10.1016/S2468-1253(22)00004-8
Roberts EA, Schilsky ML. Current and emerging issues in Wilson’s disease. New Engl J Med. 2023;389(10):922-938. doi: 10.1056/NEJMra1903585.
Członkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, et al. Wilson disease. Nat Rev Dis Primers. 2018;4(1):21. doi: 10.1038/s41572-018-0018-3.
Espinos C, Ferenci P. Are the new genetic tools for diagnosis of Wilson's disease helpful in clinical practice? J Hepatol. 2020;2(4):100114. doi: 10.1016/j.jhepr.2020.100114.
Walshe JM. History of Wilson disease: a personal account. Handb Clin Neurol. 2017; 142:1-5. doi: 10.1016/B978-0-444-63625-6.00001-X.
Dooley JS, Purchase R. History of Wilson disease. In: Weiss K, Schilsky M, editors. Wilson disease: pathogenesis, molecular mechanisms, diagnosis, treatment and monitoring. London: Elsevier; 2019. P. 3-14.
Park H, Park DK, Kim MS, Yoon JH. Pseudo-dominant inheritance in Wilson's disease. Neurol Sci. 2016 Jan;37(1):153-155. doi: 10.1007/s10072-015-2394-8.
Coffey AJ, Durkie M, Hague S, et al. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2013;136(Pt5):1476-1487. doi: 10.1093/brain/awt035.
Schilsky ML, Roberts EA, Bronstein JM, et al. A multidisciplinary approach to the diagnosis and management of Wilson disease: 2022 practice guidance on Wilson disease from the American Association for the Study of Liver Diseases. Hepatology. 2022; 00:1-49. doi: 10.1002/hep.32801.
Nagral A, Sarma SM, Matthai J, Kukkle LP, Devarbhavi H, Sinha S, et al. Wilson’s disease: Clinical Practice Guidelines of the Indian National Association for Study of the Liver, the Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition, and the Movement Disorders Society of India. J Clin Exp Hepatol. 2019;9(1):74-98. doi: 10.1016/j.jceh.2018.08.009.
Li H, Liu L, Li Y, He S, Liu Y, Li J, et al. Familial screening of children with Wilson disease: the necessity of screening in previous generation and screening methods. Medicine. 2018;97(27): e11405. doi: 10.1097/MD.0000000000011405.
Socha P, Janczyk W, Dhawan A, Baumann U, D’Antiga L, et al. Wilson’s disease in children: a position paper by the hepatology committee of the European Society for Paediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2018;66(2):334-344. doi: 10.1097/MPG.0000000000001787.
Li H, Tao R, Liu L, Shang S. Population screening and diagnostic strategies in screening family members of Wilson’s disease. Ann Transl Med. 2019;7(Suppl 2):S59. doi: 10.21037/atm.2019.03.54.
Brunet AS, Marotte S, Guillaud O, et al. Familial screening in Wilson’s disease: think at the previous generation! J Hepatol. 2012;57(6):1394-5. doi: 10.1016/j.jhep.2012.07.011.
Lorincz MT. Neurologic Wilson's disease. Ann N Y Acad Sci. 2010; 1184:173-87. doi: 10.1111/j.1749-6632.2009.05109.x.
Dzieżyc K, Gromadzka G, Członkowska A. Wilson’s disease in consecutive generations of one family. Parkinsonism Relat Disord. 2011;17(7):577-578. doi: 10.1016/j.parkreldis.2011.04.013.
Panzer M, Viveiros A, Schaefer B, Baumgartner N, Seppi K, Djamshidian A, et al. Synonymous mutation in adenosine triphosphatase copper-transporting beta causes enhanced exon skipping in Wilson disease. Hepatol Commun. 2022;6(7):1611-1619. doi: 10.1002/hep4.1922.
Vrabelova S, Letocha O, Borsky M, Kozak L. Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Mol Genet Metab. 2005;86(1-2):277-85. doi: 10.1016/j.ymgme.2005.05.004.
Ferenci P, Stremmel W, Czlonkowska A, et al. Age, and sex but Not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology. 2019;69(4):1464-1476. doi: 10.1002/hep.30280.
Sanchez-Monteagudo A, Alvarez-Sauco M, Sastre I, Martinez-Torres I, Lupo V, Berenguer M, et al. Genetics of Wilson disease and Wilson-like phenotype in a clinical series from eastern Spain. Clin Genet. 2020;97(5):758-63. doi: 10.1111/cge.13719.
Trocello JM, Balkhi S, Woimant F, Girardot-Tinant N, Chappuis P, Lloyd C, Poupon J. Relative exchangeable copper: a promising tool for family screening in Wilson disease. Mov Disord. 2014;29(4):558-562. doi: 10.1002/mds.25763.
Woimant F, Djebrani-Oussedik N, Poujois A. New tools for Wilson’s disease diagnosis: exchangeable copper fraction. Ann Transl Med. 2019;7(Suppl 2):S70. doi: 10.21037/atm.2019.03.02.