
Introduction
Vascular endothelium (VE) plays a versatile role in general, and especially, cardiovascular homeostasis control. Since the discovery of nitric oxide (NO) as an endothelium derived factor having a key role in vascular tone regulation, a lot of data are still accumulated available to depict a multi-facet caliber of VE. To note in this regard the property of VE to prevent platelet adhesion and prothrombotic state activation, circulating white cells passage, pathological vascular remodeling and atherogenic plaque progressing [1-3]. Likewise, VE influences angiogenesis and arteriogenesis, processes that have a dichotomic significance, meaning they can have both positive and negative effects, the last being touchy linked to cancer growth and cancer spreading [4].
The VE functions in veins and arteries are basically common, although some differences exist, such as: (i) venous endotheliocytes express more amount of NO; (ii) endothelial junctions in arteries appears to be tighter; (iii) expression of endothelial vascular growth factor (VEGF) and of receptors against von Willebrand factor is higher in the endotheliocytes of arteries [5]. Remarkably, large veins versus arteries have a greater capacity to trigger and sustain an inflammatory response. Likewise, in veins the formation of thrombus is much faster than in arteries due to lowered blood flow, and the red blood thrombus does not need von Willebrand factor.
Despite the fact that both arteries and veins express a lot of common markers, the genes families inherent to endotheliocytes of arteries and veins are not totally similar. Arterial endotheliocytes express more opulently ephrinB2 gen, but in veins predominates EphB4 gen [6]. Conceptually is admitted that hemodynamic stress of the blood flow might change the phenotype and morpho-functional support of vessel behavior regardless a certain identical embryonal pattern. Thus, the increased blood flux can induce an arterial phenotype of venous endothelial cells.
Noteworthy, the VE injury associated by various models of dysfunction becomes an important pathogenic pillar of many homeostasis disorders, which in veins are mostly manifested by high risk of thrombus formation (e.g. deep venous thrombosis), and in arteries by vascular remodeling developing in a field of atherogenic event and artery reactivity disturbances. Artery endothelial dysfunction is viewed as a pivotal tool of so dangerous cardiovascular disorders like acute myocardial infarction, stroke, and arterial hypertension. Endothelial damage associating the coronary angioplasty triggers the process of in-stent re-stenosis qualified as a pattern of pathological vascular remodeling resulting in the angioplasty benefic lost.
Taken together these arrangements underline the real importance of early endothelial dysfunction detection for prediction of cardiovascular homeostasis impairment as well as for disentangle of main therapeutic targets. Therefore, a multi-marker strategy or multi-marker panel is applied as a feasible algorithm containing markers which are referring to most important pathogenic interfaces of the endothelial dysfunction and its imminent pathological consequences.
Material and methods
In order to build this review article, the searching of needed references concerning actuality, specificity, relevance and matching to article goal was projected on databases of PubMed, MEDLINE Google Scholar and Cochrane with the depth of the relating up to the year 2000. The found articles were structured in regard to main objectives, comprehensively analyzed, and the principal coagulated entities have been critically exposed.
Results and discussions
Conceptually the multi-marker algorithm is built from a puzzle of various pathogenic mechanisms leading to VE injury and dysfunction whose most important expression is NO lack. In arteries these phenomena are closely linked to atherogenesis and inducing endogenous and exogenous factors. Leader mechanisms taken as intelligible objectives for seeking feasible markers and predictors of VE dysfunction and its repercussions are inflammation, oxidative stress, endothelial reendothelization potency, decreases expression and activity of endothelial nitric oxide synthase (eNOS), smooth vascular myocyte migration and proliferation control disturbance, hemostasis disorders, etc.
Inflammation and more important circulating markers
Chronic or low-grade systemic inflammation or subclinical inflammation is considered as a key factor leading to VE impairment and dysfunction, beyond its direct action on atherogenesis progressing. Contemporary concept corroborates chronic inflammation as a sustained elevation of circulating cytokines released by a diversity group of cells, including adipocytes, which promotes an impact on remote endothelial cells. Classical canon underlines obesity, metabolic syndrome and insulin resistance as most important pathological entities leading to subclinical inflammation even in both relatively young people and apparently healthy adults [7-9].
Pathophysiological interface of inflammation induced VE disorders is very complex, multi hierarchical and interdependent [10, 11]. Among most important mechanisms should be revealed:
Inflammatory cytokines induced endothelial cell apoptosis and pyroptosis.
Activation of oxidative stress.
Reduced expression and activity of eNOS.
Diminution of tetrahydrobiopterin, a cofactor of NO synthesis by endothelial cells from L-arginine.
Increased activity of arginase, which takes supplementary amounts of L-arginine from eNOS-citrulline cycle and converts this amino acid in ornithine cycle leading to urea formation [12]. Remarkably, arginase II expression in mitochondrial apparatus is augmented by inflammatory cytokines released by proinflammatory macrophages type 1 (M1). On the other hand, excess of polyamines released in ornithine cycle triggered by arginase are able to increase production of asymmetric dimethylarginine which inhibits activity of eNOS [13].
Inflammation boosting resulting in endothelial dysfunction is higher in arterial vessels due to increased risk of disturbed blood flow. Contrarily, laminar blood flow like flow in veins is not a so strong factor capable to trigger inflammatory response, and as consequence it means a lowered hazard for excessive synthesis of ROS, reactive oxygen species (figure 1).
|
Fig 1. Mutual relation ROS-inflammation is sustained by disturbed blood flow [11] |
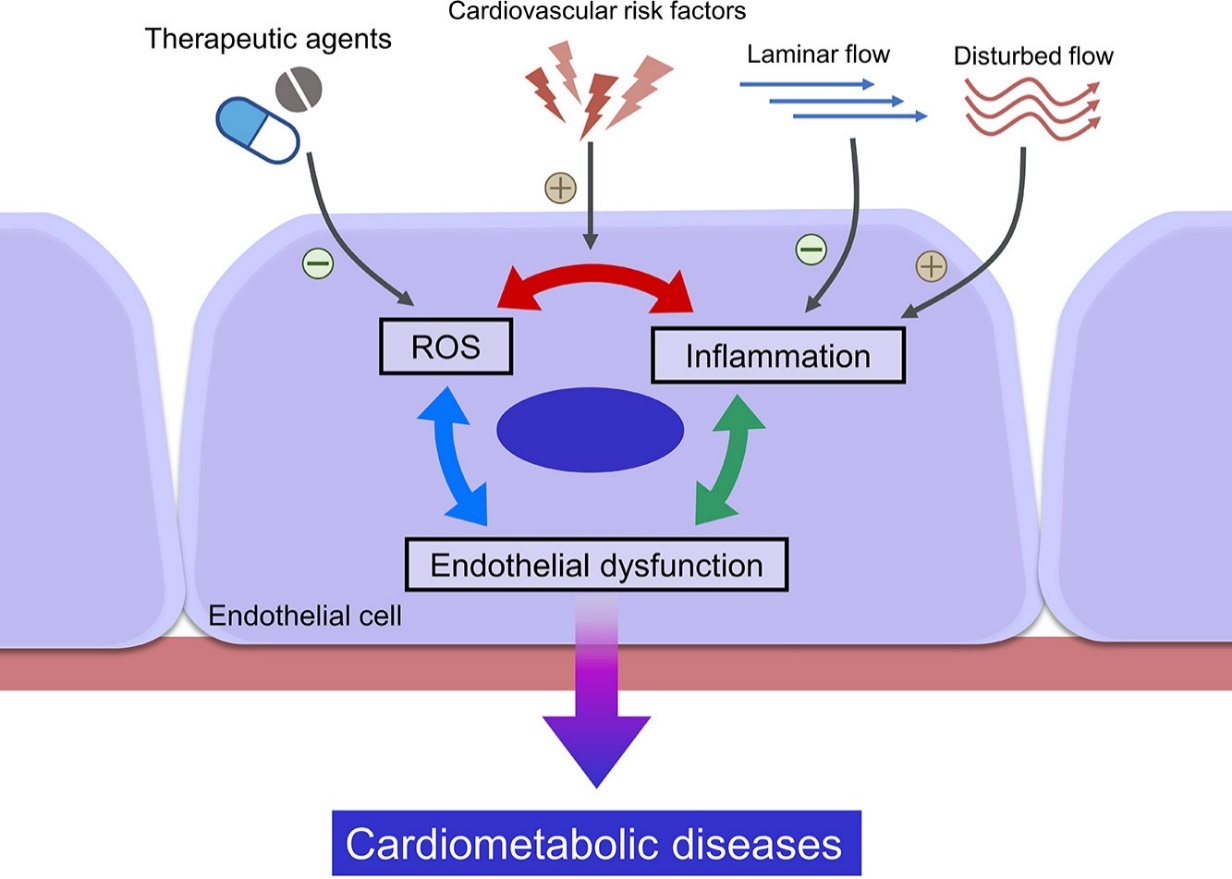
Finally, inflammation either directly or indirectly (thereby of NO deficiency) exacerbates the process of atherosclerosis a one of crucial supports responsible for VE damage and dysfunction. Inflammation also is a factor promoting vascular ageing either of arteries or veins which strongly correlates with VE dysfunction severity [14].
Accordingly, the assessment of inflammatory markers is an important diagnostical tool of VE dysfunction and respectively a prediction of inherent cardiovascular disorders. The most frequently used marker of inflammation and VE dysfunction is high-sensitive C-reactive protein (hsCRP), which compresses a large amount of evidence. Likewise, hsCRP is considered a key predictor of coronary and heart failure risk [15, 16]. To note in this regard that the serum level of hsCRP is taken as cardiovascular risk stratification such as the levels of <1 mg/L, 1–3 mg/L, and >3 mg/L should be interpreted respectively as low, moderate, and high vascular risk, respectively, based on a large number of population studies [17]. Must be mentioned that elevation of serum hsCRP level more than 15 mg/L should be interpreted as a presence of pathogen induced inflammatory processes. Furthermore, moderate increased hsCRP levels should be taken into consideration in cases when where are other cardiovascular risk factors like arterial hypertension, diabetes, obesity, smoking, dyslipidemia, stress, etc. [18].
The CRP role in endothelium damage and dysfunction is well documented in an enormous number of clinical and experimental studies and is based on certain mechanisms.
First, CRP is early and tightly involved in triggering and progressing of atherosclerosis in arteries and thrombosis in veins (figure 2).
|
Fig 2. Synoptic mechanisms of pentameric CRP (pCRP) and monomeric CRP (mCRP) involvement in the processes of atherosclerosis in arteries and thrombosis in veins [19]. |
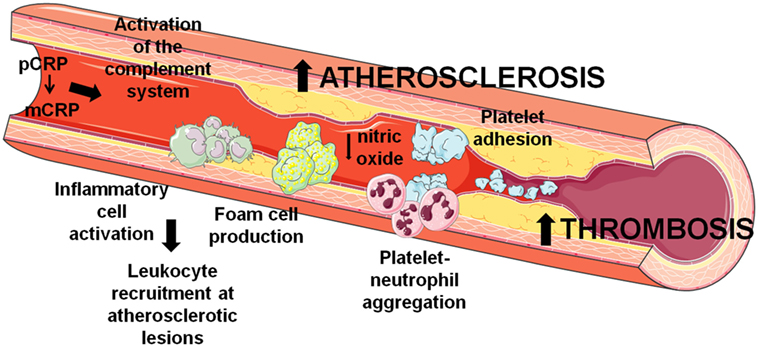
The main mechanisms of CRP induced inflammation driving VE dysfunction are linked to complement activation, stimulation of foam cell formation and production of ROS and cytokines, circulating leukocyte recruitment, stimulation of platelet adhesion and aggregation, inhibition of eNOS and NO diminution [19, 20]. More than that, the amount of CRP found in atherogenic plaque robustly correlates with intensity of inflammatory response, endothelial dysfunction degree and risk power regarding acute coronary syndrome. In regard to the ratio of monomeric/pentameric CRP forms in veins and arteries is to underline the role of mechanical and physical characteristics of the blood stream which determine the activity of pentamer conversion in monomer as well as the enzyme activity of the vascular locus. It is considered that in arteries the concentration of monomeric CRP is bigger than in veins, primarily due to a stronger blood stream, and a higher proteolytic enzyme concentration especially in the region of atherosclerotic injury [21]. It is still unknown the precise mechanisms explaining more aggressive pro-atherosclerotic action of CRP monomers in comparison to CRP pentamers although both structural patterns activate the same pathophysiological events, such as complement activation, platelet and leukocyte adhesion, endothelial cells damage [22]. Moreover, in the arterial endothelial zones of injury the concentration of white blood cells is higher compared to veins, a fact which should be taken into consideration, because according to some opinions, lymphocytes can synthesize CRP monomers [23]. Respectively, the CRP-linked and mediated inflammatory process is significantly more active in arteries than veins. Endothelial inflammation is manifested by increased expression of phospholipase A2 which facilitates the pCRP entering in the liposomes of cell membrane (e.g., endotheliocytes, macrophages, smooth muscle cells) where it is exposed to a process of dissociation in partly due to an acidic microenvironment. So, in a case of a certain suspected risk to endothelial dysfunction both pCRP and mCRP circulating levels are diagnostically more important for arteries.
Second, CRP increases expression of monocyte chemoattractant protein (MCP-1), as well as of selectins in endothelium [24]. These events facilitate the monocyte and neutrophil traffic through endothelial layer. The trapped white cells trigger the process of smooth myocytes migration and proliferation not only in arteries, but also in veins, defined as vein wall remodeling [25]. It is sustained by extracellular matrix (ECM) excessive degradation and synthesis under the action of fibroblasts and ECM metalloproteinases. Noteworthy that under statins action, the serum MCP-1 content decreased and it resulted in improvement varicose vein remodeling [26]. Likewise, it is a corroborated suggestion that susceptibility of the vein ECM to proinflammatory agents’ action is linked to some proteomic and genetic disorders, which justifies the application of multi-marker panel in order to unravel the real predictors of chronic venous disease [27].
Third, CRP decreases activity of HDL capacity to take cholesterol from foam cells leading to atherogenic phenomenon boosting. On the other hand, HDL markedly reduces the proinflammatory activity of CRP, and therefore lipid profile improvement manifested by LDL decrease and HDL raise confines the pathogenic approach of CRP in inflammation induced VE damage and dysfunction [28]. This effect is especially suitable in arteries and represents a pillar of lipid-lowering therapy.
Another remarkable marker of inflammation and predictor of VE dysfunction is tumor necrosis factor alpha (TNF-α). TNF-α is considered as a pivotal proinflammatory marker whose expression is dependent on activity of 2 transcription factors: nuclear factor kappa B (NF-kB) and nuclear factor of activated T lymphocytes (NFAT) which provide the signals from membrane Toll-like receptors to nuclear DNA. The strong contribution of TNF-α in endothelial inflammation and dysfunction was confirmed in a lot of clinical and fundamental studies. Especially are the emphasized results showing that intra-arterial TNF-α infusion in healthy volunteers resulted in acute vascular inflammation associated with impaired endothelium-dependent vasorelaxation [29].
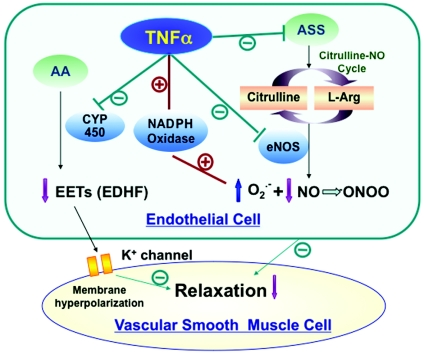
A few important detrimental effects of TNF-α are recognized as (figure 3):
Increased activity of endothelial membrane NADPH-oxidase leading to excessive formation of superoxide anion which neutralizes the NO by accumulation of peroxynitrite (ONOO-), a radical able to constrict arteries and exacerbate atherosclerotic process.
Inhibition of cytochrome 450 resulting in lowering of epoxyeicosatrienoic acids (EETs) formation from arachidonic acid, which is able to relax arteries by hyperpolarization mechanism.
Decreased expression of eNOS.
|
Fig 3. Mechanisms of TNF-α induced endothelial dysfunction [29]. AA – arachidonic acid; EDHF – endothelial derived hyperpolarizing factor; L-Arg -L arginine. |
The circulating TNF-α levels directly correlate with severity of endothelial dysfunction in both arteries and veins. In some cases, overexpression of TNF-α is linked to genetic disorders found in autoimmune diseases (e.g., rheumatoid arthritis, systemic lupus erythematosus) associated with VE dysfunction worsening [30]. TNF-α stimulates expression of endothelial receptor LOX-1 which boosts sequestration of oxiLDL molecules from blood in order to build the atherogenic plaque in arteries. LOX-1 activation also leads to increased expression of NF-kB and arginase2. The last reduces NO formation because consumes excessively L-arginase in the ornithine cycle.
The impact of low grade of inflammation or subclinical inflammation often found in diverse metabolic disorders (e.g., obesity, diabetes, insulin resistance syndrome) on VE even in young persons is provided via elevated circulating levels of TNF-α. Impairment of microcirculation (cerebral, cardiac), a system of vascular network which encompasses resistant vessels such as prearterioles, arterioles and capillaries, having a risk power for stroke and acute myocardial infarction, is strongly linked to TNF-α elevation [31]. Being directly and actively involved in the ROS formation, TNF-α leads to endothelial cell activation in order to release endothelin 1 (ET-1) which is considered as one of most potent natural vasoconstricting agents. So, collectively TNF-α, ROS and ET-1 are important pathogenic factors acting together and able to induce VE dysfunction and promote its consequences. More than that, TNF-α increases expression of ET-1 receptor in smooth arterial myocytes (i.e., types ETA and ETB), thus tightly contributing to artery remodeling, especially resistant arteries [32]. Stimulation of ET-1 generation by TNF-α is mediated by c-Jun N-terminal kinase pathway, which is also a mechanism of ROS release. Perivascular adipose tissue is influenced via inherent adipokines the rate of TNF-α expression. It has been proven that adiponectin decrease provides TNF-α expression, but in excess leptin and resistin, in contrary, stimulates expression. The substance P can decrease in natural conditions the TNF-α impact on VE thereby a mechanism linked to modulation of proteinkinase B (Akt) triggering eNOS activity [33].
Recent studies have shown that diverse families of micro-ARN (miR) can have a dichotomic manner of VE regulation. In this regard is important the action of mir-29a-3p manifested by decrease of TNF-α receptor expression, and as consequence the TNF-α induced activation of adhesive molecules, such as E-selectin and ICAM-1 (intercellular adhesion molecule) and VACM-1 (vascular adhesion molecule), hence playing a role of natural VE protector and a mechanism counteracting endothelial dysfunction worsening [34].
The role of TNF-α in the pathogenesis of inflammation induced and assisted endothelial injury is quite well documented [35]. However, the predictive power of TNF-α regarding early endothelial dysfunction is better studied and known for arterial bed.
Both main markers of VE dysfunction, CRP and TNF-α, are conceptually strongly linked to another proinflammatory marker, IL-6. Inflammation, oxidative stress, and Ang II stimulate the IL-6 expression in veins and arteries wall. Being a multifunctional cytokine, IL-6 acts on diverse cells (endotheliocytes, myocytes, adipocytes, monocytes, and cardiomyocytes) in both endocrine and paracrine pathways. Like CRP, IL-6 can be synthesized locally, in the atherosclerotic plaque, and its level positively correlates with expression of LOX-1 receptors and pathological pattern of vascular remodeling. IL-6 receptor is found in two arrangements [36]:
As a membrane receptor without intrinsic kinase activity and with low affinity at the cell surface.
As a gp130 transmembrane site having intrinsic tyrosine kinase activity able to bind the circulating complex acting as a ligand: IL-6+ soluble IL-6 receptor.
Activation of both markers leads to activation of STAT-1 and STAT-3 (signal transducer and activator of transcription) and consequently to nuclear DNA activation. The gp130 is expressed ubiquitously, but membrane IL-6 receptor – selectively, in dependence of the cell type.
In a comparative analysis of IL-6 role vis-à-vis of venous and arterial endothelial dysfunction it is important to underline 2 distinct traits. The first, vascular wall capacity to release IL-6 is higher in arteries. Second, the gp130 expression level in veins is lowered.
Among pleiotropic effects of IL-6 should be highlighted the following actions in regard to its role in VE dysfunction [37, 38]:
augments the detrimental action of TNF-α and CRP on VE;
increases production of ROS;
stimulates expression of chemokines;
elevates the level of plasmin activator inhibitor;
stimulates synthesis of active-phase proteins by liver (e.g., fibrinogen, ceruloplasmin);
increases expression of adhesive molecules and facilitates the trans-endothelial traffic of white blood cells;
triggers the migration and proliferation of endothelial cells and smooth vascular muscle cells;
activates the ECM fibroblasts and promotes the vascular remodeling.
According to IL-6 intake in the venous endothelial dysfunction the majority of evidence indicate that in diverse patterns of vein remodeling the circulating IL-6 level is significantly elevated and robustly correlates with the serum amount of IL-8, TNF-α and MCP-1 [39, 40]. Anyway, still the amplitude of diagnostic and pathophysiological relevance of the main proinflammatory markers such as CRP, TNF-α and IL-6 in veins remains less appreciated in comparison with arteries. First and foremost, deep venous thrombosis remains the principal venous pathology requesting monitoring of inflammation markers as predictors of endothelial dysfunction and inherent repercussions.
Markers of endothelial lesion and reendothelization
Endothelial lesion is a continuous process triggered by various pathologic factors such as: ROS, inflammation mediators, shear-stress, hyperglycemia, dyslipidemia, hyperhomocisteinemia etc. Many comorbidities (e.g., arterial hypertension, diabetes, autoimmune diseases) worsen endothelial injury leading to a progressive decline of VE dysfunction. The most important markers of endothelial lesion are endothelial microparticles (EMP), endothelial exosomes which derive from special vesicles erupted from endothelial cell membrane and endothelial apoptotic bodies [41-43]. The circulating level of EMP positively correlates with the degree of endothelial injury especially in arterial bed, atherosclerotic process activity as well as risk of acute vascular accidents like acute myocardial infarction and stroke. Likewise, elevated EMP level is associated with higher serum amounts of CRP, TNF-α, IL-6, IL-8, MCP-1, and diabetes induced VE damage [44]. Hyperhomocysteinemia, smoking and hypodynamic lifestyle are often associated with raised EMP and free endotheliocyte circulation that indicates on endothelial layer injury and endothelial inflammation because the level of phospholipase A2 is also increased.
In the venous system, the pathophysiological significance of EMP is rather linked to procoagulant activity and risk of thrombosis, and this marker predicts endothelial cell senescence and functional weariness [45]. To note in this context that EMP increases directly and indirectly expression of selectins, VACM-1 and ICAM-1, activates platelets and monocytes, increases release of tissue factor from endotheliocytes, thrombocytes and mononuclear cells. These procoagulant events are also associated with elevated levels of thromboxane A2 (TxA2), ET-1, ROS and peroxynitrite, growth factors and plasminogen activator inhibitor. So, EMP is an early predictor of vein thrombosis, and obviously this marker should be assessed in association with other biomarkers referring to coagulant, anticoagulant and fibrinolysis systems feasibility.
The process of reendothelization is a crucial phenomenon aiming substitution of either damaged cells or senescence cells by new endotheliocytes. In regard to renewing of senescence cells population, should be emphasized that both endotheliocytes and smooth vascular myocytes demonstrate an advanced statement of senescence in the boosted atherosclerotic process [46]. Endothelial senescence and damage lead to diminution of tetrahydrobiopterin, a cofactor of NO synthesis, and respectively a predictor of VE dysfunction. Therefore, tetrahydrobiopterin became a reliable marker of early VE dysfunction and atherosclerosis progression [47].
Basically, reendothelization is realized and emphasized by following paramount markers: VEGF, angiopoietins, and endothelial progenitor cells (EPCs).
VEGF is considered a strong endotheliocyte mitogen and angiogenic factor. In vitro studies have demonstrated that VEGF stimulates the growth of arterial, venous, and lymphatic endotheliocytes which are forming a new capillary network. Hypoxia, ischemia, and oxidative stress are main factors triggering expression of VEGF, which is underlined also as vasoprotector factor due to its antiapoptotic effects derived from activation of Bcl-2 proteins. Mechanical stress of VE induced by diverse maneuvers of angioplasty damages endothelial cells and stimulates release of VEGF. Nitric oxide and eNOS play an important role in the VEGF induced angiogenesis [48]. Remarkably, VEGF induces vasodilation in a dose dependent manner, and this effect is fought to be mediated by prostacyclin. Two types of receptors mediate the large spectrum of VEFG effects: R1 and R2. The VEGF-R2 receptor is expressed on arterial and venous vasculature, being involved in control of vasodilation, atherosclerosis, cell migration and proliferation [49].
VEGF-R1 receptor is mostly found as a soluble receptor capable to bind the circulating VEGF and the affinity of receptor against specific ligand is 10-fold higher versus VEGF-R2. However, the kinase activity triggered by via VEGF-R1 is 10-fold weaker [50]. In contrast to VEGF-R2, VEGF-R1 does nor mediate angiogenesis in embryonal tissue. In adulthood it is expressed in both endothelial cells and macrophages and worsens the atherosclerotic process.
Accumulated data suggest that VEGF is a predictor of arterial wall remodeling, and its elevated level is well proven in patients with arterial hypertension and type 2 diabetes [51]. The authentic role of VEGF in reendothelization was confirmed in vivo by administration of an antibody neutralizing this growth factor after angioplasty induced vascular injury, which led to VE recovery annihilation. It has been deduced that released VEGF from damaged endothelial cells in the blood promotes the function of EPCs, which are sequestrated from bone marrow under action of this growth factor. Increased production of endogenous NO after an adequate reendothelization realized basically by EPCs, decrease expression and activity of VEGF. More than that, exogenous NO released from nitrite donors reduces the activity of reendothelization in vivo due to decrease of VEGF level as well as level of APCs. Interestingly, carbon monoxide, another endothelial derived factor, acts unlike NO, contributing to VEGF expression increase thereby hypoxia inducible factor. Finally, VEGF being appreciated as a growth factor expressed not only by endotheliocytes (e.g., by macrophages, platelets, keratinocytes, renal mesangial cells) plays a certain role in other functions besides reendothelization, such as: hematopoiesis, wound healing, bone tissue synthesis. To be noted in this context that tumor cells also are capable to express VEGF, and angiogenesis becomes a pathogenic factor facilitating tumor growing and metastasis [52]. Likewise, use of the blockers of VEGF receptors led to a suppressing effect on tumor growth and lowered risk of tumor metastasis.
Thus, EPCs are proven as an important tool triggering and sustaining reendothelization because they are capable to differentiate into endothelial cells and hence provide phenomenon of new blood vessels formation. Therefore, EPCs are often named as circulating angiogenic cells. Mobilization of EPCs from bone marrow is realized not only by VEGF, but other factors are also available, such as ECM metalloproteinase 9 and stromal cell-derived factor 1. Also, EPCs can secrete some paracrine factors, such as IL-8 and stromal cell-derived factor 1. Hypoxia and ischemia are factors that mobilize EPCs in blood flow resulting in their migration toward the zone of endothelial injury where they proliferate, and differentiate into mature endothelium, thereby leading to reendothelialization and neovascularization [53, 54]. Being injected intravenously EPCs can reach the infarcted region within 48 hours.
Decreased levels of EPCs or their functional disability are strong predictors of endothelial dysfunction and cardiovascular disease as well [55]. Lower levels of EPCs are associated with a weaker process of reendothelization even when the circulating level of VEGF is quite high. It is a well proven fact, that low levels of EPCs are accompanied by decreased production of NO and vascular reactivity impairment. Weak sequestration of EPCs from bone marrow augments VE dysfunction and accelerates the progression of cardiovascular disorders. Many cardiovascular risk factors confine the activity of EPCs recruitment and their home in the zone of VE injury, such as hyperglycemia, hypercholesterolemia, hyperhomocysteinemia, low grade inflammation, leukocytosis, oxidative stress, etc. Therefore, for a better understanding of real pathogenic interface of VE dysfunction, EPCs should be assessed together and correlatively with a lot of other markers and risk factors of vascular, especially arterial damage. Noteworthy, the level of circulating EPCs is recognized as an independent predictor of atherosclerosis progression and suspected disorders of artery remodeling and reactivity [56-58].
Nowadays the serum concentration of EPCs is viewed as an important diagnostic marker as well as a therapeutic target of cardiovascular disorders associated with VE dysfunction. Intracoronary infusion of EPCs lead to reduced myocardial infarction zone and to improvement of myocardial and ECM remodeling in post-infarction period.
In this regard it is important the opinion of K. Lenk et al. (2021) who suggest that EPCs are a one of key tools for providing physical exercise benefits on atherosclerosis and coronary disorders [59]. Physical activity maintains the structural integrity of VE regardless of any risk factors action, and in case of endothelium alteration, the level of EPCs is higher and their involvement in vascular repair becomes more efficient.
Regarding some differences of EPCs role in arteries and veins, there is no conclusive data. Nevertheless, could be relevant hypothesis that the EPCs role in both arterial and venous endothelium repair is the same.
Nowadays, a new marker of reendothelization is angiopoietin which belongs to group of growth factors. Two most important families of angiopoietin are known: Ang-1 and Ang-2, which are natural ligands of the Tie-1 and, respectively, Tie-2 receptors tyrosine kinase, which are expressed primarily on endothelial cells and early hematopoietic cells [60, 61].
Ang-1 acts as a potent angiogenic growth factor, but Ang-2 plays an important role in various physiological processes and its impairment is inherent to a lot of homeostasis disorders, including for the lymphatic system.
Tie-2 receptor is abundantly expressed in endothelium, especially of arteries. Likewise, vascular fibroblasts can express Tie-2 receptors, whose attribution is considered to be tightly linked to vascular injury repair. Therefore, this receptor is considered as atheroprotective in arterial endothelium [62]. So, both angiopoietins fulfill a lot of suitable functions, such as: adhesion and survival of endothelial cells, augmentation of EPCs action vis-à-vis of endothelium repair, reendothelization and angiogenesis from new formed capillaries. However, the authentic equilibrium between Ang-1 and Ang-2 in postnatal vascular morphology and physiology control is still not well established. Most hypotheses state that Ang-1 acts in a paracrine agonistic manner inducing Tie-2 phosphorylation and subsequent vessel stabilization. In contrast, Ang-2 is produced by endothelial cells and acts as an autocrine antagonist of Ang-1-mediated Tie2 activation. Conceptually is important that Ang-2 blunts the action of proinflammatory cytokines on vascular endothelium and therefore prevents and mitigates the VE injury and remodeling. Lastly, Ang-2 and EPCs action on VE might be boosted by TGF-beta (transforming growth factor) whose signal is received by a special endothelial receptor, endoglin (membrane glycoprotein) whose activation promotes the neoformation of capillaries and integrity of vessel walls, either in the embryo or postnatal life [63].
Together with VEGF, angiopoietins could stimulate angiogenesis induced tumor metastasis, and respectively are depicted as targets of therapy, especially concerning the Ang-2. More data is needed in order to highlight physiological and pathophysiological entities of these growth factors regarding VE dysfunction.
Markers of VE dysfunction in connection to hemostasis disorders
Vascular endothelium dysfunction is associated with severe hemostasis disorders manifested finally by a prothrombotic state induction. Accordingly, VE dysfunction becomes a condition of prothrombotic risk, but at the same time the formed thrombi comprise a pathogenic interface for DE dysfunction exacerbation. Therefore, inherent markers of hemostasis disorders should be important and significant predictors of VE dysfunction severity and its prognostic outcomes.
Basically, prothrombotic state is a result of either overactivation of coagulation system or impairment of anticoagulant processes. Frequently these factors act together. Fibrinolytic activity of the blood also plays a notable role because can ensure in time the resolution of fibrin thrombus. Endothelium incompetency in the field of hemostasis control is emphasized as following main entities:
Discovery of the integrins receptors expressed by subendothelial collagen fibers.
Platelet activation and increase of its adhesive and aggregation receptors.
Increased release of von Willebrand factor (vWF).
Excessive accumulation of the tissue factor released in partly from damaged endotheliocytes.
Decreased expression of endothelial receptors playing a crucial role in the anticoagulant protein C activation.
Diminution of the anti-thrombin III level.
Reduced NO and prostacyclin production.
The earliest consequences of VE lesion and dysfunction in regard to hemostasis control are elevated circulating level of vWF, antithrombin III and incompetence of anticoagulant tandem protein C-protein S. Von Willebrand factor, a pentameric glycoprotein, is mainly synthesized by endothelial cells (megakariocytes also express vWF) and is stored in Weibel-Palade bodies and a-granules respectively of endotheliocytes. Normally the circulating level of vWF is linked to ABO blood groups and other genetic arrangements. Non-genetic background of vWF plasma level changes is determined by age, gender, inflammation, oxidative stress, and surely by endothelial cell integrity in either arteries or veins. Most important hemostatic functions of vWF are driven by: (i) stabilization of the factor VIII in the circulation because vWF serves as its plasma carrier, and (ii) boosting of platelet adhesion to vascular wall and platelet aggregation, respectively.
Majority of pathologic states associated with vascular endothelium injury and dysfunction demonstrate elevated circulating levels of vWF [64-66]. Increased plasma vWF concentration means a risk for prothrombotic state activation leading to thrombi formation in arteries of veins (so, white and red thrombi). Most important conceptual and practice question remains as: does the vWF has a more decisive role in thrombus formation in arteries or in veins?
Solitary narrations indicate the importance of discovery of all active sites of vWF pentamer in the blood flow in order to achieve maximal hemostatic functions [67-69]. This is possible in an arterial rapid and intense blood flow comparable to the slow flow in. So, the predictive power of increased plasma level of vWF concerning risk of thrombus formation is obvious in arteries. In regard to vWF role in venous thrombus it is linked to lowered polymer degradation and consequently less fully discovery of active sites of glycoprotein needed for factor VIII binding due to weak blood flow in veins [70].
Another hemostatic factor linked to endothelial availability is antithrombin III (AT-III), recognized as an endogenous serine protease inhibitor (glycoprotein consisting of 432 amino acid residues), thus of thrombin. Endothelial cells injury leads to decreased release of AT-III resulting in a lowered capacity of thrombin inactivation of the blood. Likewise, AT-III also inhibits other factors of coagulation system, such as IX, X, XI and XII [71].
So, blood levels of AT-III predicts VE dysfunction and risk of serious cardiovascular diseases like acute myocardial infarction and stroke [72].
Finally, anticoagulant protein C is a key antithrombotic factor, but its functional feasibility is closely linked to endothelial receptor (type 1 transmembrane glycoprotein) needed for protein C activation. Therefore, even the normal or elevated circulating levels of protein C could be inefficient to prevent thrombus formation if due to endothelial damage its activation is compromised. However, there are interesting approaches to study the predictive value of soluble endothelial receptors to protein C. Remarkably, in the blood stream this receptor binds to circulating protein C, but this does not result in anticoagulant factor activation, but in contrary leads to its inhibition due to lost ability to inhibit factor Va [73].
Thus, increased level of soluble endothelial receptor of protein C could be a reliable marker of anticoagulant capacity fall due to protein C misfunctioning, and respectively a predictor of prothrombotic state activation in both arteries and veins.
Conclusions
The multi-marker panel of endothelial dysfunction is a key opportunity to identify the markers referring to main pathogenic mechanisms, such as inflammation, reendothelization and hemostasis disorders having a reliable early prediction for arterial and venous endothelial dysfunction.
Although majority of markers have a same predictive value of endothelial dysfunction in both arteries and veins, the circulating levels of CRP and vWF are more important in regard to arteries because here depolymerization activity of these pentamers is higher leading to pathogenic mission augmentation concerning vascular remodeling and prothrombosis.
The circulating level of protein C is not a reliable marker of endothelial dysfunction induced risk of thrombosis. However, the elevation of its soluble endothelial receptor could predict lower anticoagulant activity of protein C due to the loss of capacity to inactivate factor Va.
Competing interests
None declared.
Authors’ contribution
Both authors contributed equally to the literature searching, conceptual highlighting of the material as well as writing of the manuscript. The authors read and approved the final version of the manuscript.
Authors’ ORCID IDs
Victor Ojog – https://orcid.org/0000-0002-2386-6654
Svetlana Lozovanu – https://orcid.org/0000-0002-5777-1805
References
Stanek A, Fazeli B, Bartus S, et al. The role of endothelium in physiological and pathological states: new data. BioMed Res Int. 2018;2018:1098039. doi: 10.1155/2018/1098039.
Bkaily G, Jacques D. Morphological and functional remodeling of vascular endothelium in cardiovascular diseases. Int J Mol Sci. 2023;234(3):1998. doi: 10.3390/ijms24031998.
Campinho P, Vilfan A, Vermot J. Blood flow forces in shaping the vascular system: a focus on endothelial cell behavior. Front Physiol. 2020;11:552. doi: 10.3389/fphys.2020.00552.
Heil M, Eitenmuller I, Schmitz-Rixen T, Schaper W. Arteriogenesis versus angiogenesis: similarities and differences. J Cell Mol Med. 2006;10(1):45-55. doi: 10.1111/j.1582-4934.2006.tb00290.x.
Niklason L, Dai G. Arterial venous differentiation for vascular bioengineering. Ann Rev Biomed Eng. 2018;20:431-447. doi: 10.1146/annurev-bioeng-062117-121231.
Wolf K, Hu H, Isaji T, Dardic A. Molecular identity of arteries, veins, and lymphatics. J Vasc Surg. 2019;69:253-262. doi: 10.1016/j.jvs.2018.06.195.
Skrzypczyk P, Ozimel A, Ofiara A, et al. Markers of endothelial injury and subclinical inflammation in children and adolescents with primary hypertension. Centr Eur J Immunol. 2019;44(3):253-261. doi: 10.5114/ceji.2019.89597.
Virdis A, Masi S, Colucci R, et al. Microvascular endothelial dysfunction in patients with obesity. Curr Hypertens Rep. 2019;21:32. doi: 10.1007/s11906-019-0930-2.
Cowan S, Leeming E, Sinclair A, et al. Effect of whole foods and dietary patterns on markers of subclinical inflammation in weight-stable overweight and obese adults: a systematic review. Nutr Rev. 2019;78(1):19-38. doi: 10.1093/nutrit/nuz030.
Theofilis P, Sagris M, Oikolomou E, et al. Inflammatory mechanisms contributing to endothelial dysfunction. Biomedicines. 2021;9(7):781. doi: 10.3390/biomedicines9070781.
Wang L, Cheng C, Yi M, et al. Targeting endothelial dysfunction and inflammation. J Mol Cell Cardiol. 2022;168:58-67. doi: 10.1016/j.yjmcc.2022.04.011.
Rath M, Muller I, Kropf P, et al. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532. doi: 10.3389/fimmu.2014.00532.
Mangoni A, Tommasi S, Sotgia S, et al. Asymmetric dimethylarginine: a key player in the pathophysiology of endothelial dysfunction, vascular inflammation, and atherosclerosis in rheumatoid arthritis? Curr Pharm Des. 2021;27(18):2131-40. doi: 10.2174/1381612827666210106144247.
Lee MKY, Vanhoutte PM. Inflammation and endothelial dysfunction with aging. In: Dauphinee S, Karsan A, editors. Endothelial dysfunction and inflammation. Basel: Springer; 2010. https://doi.org/10.1007/978-3-0346-0168-9_11.
Maio R, Perticone M, Suraci E, et al. Endothelial dysfunction and C-reactive protein predict the incidence of heart failure in hypertensive patients. ESC Heart Fail. 2021;8(1):399-407. doi: 10.1002/ehf2.13088.
Medina-Leyte DJ, Zepeda-Garcia O, Domingues-Perez M, et al. Endothelial dysfunction, inflammation and coronary artery disease: potential biomarkers and promising therapeutical approaches. Int J Med Sci. 2021;22(8):3850. doi: 10.3390/ijms22083850.
Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107(3):499-511. doi: 10.1161/01.CIR.0000052939.59093.45.
Lee HA, Choi EJ, Park B, et al. The association between metabolic components and markers of inflammatory and endothelial dysfunction in adolescents, based on the Ewha Birth and Growth Cohort Study. PLoS One. 2020;15(5):e0233469 doi: 10.1371/journal.pone.0233469.
Badimon L, Pena E, Arderiu G, et al. C-reactive protein in atherothrombosis and angiogenesis. Front Immunol. 2018;9:430. doi: 10.3389/fimmu.2018.00430.
Paffen E, Moniek P. C-reactive protein in atherosclerosis: a causal factor? Cardiovasc Res. 2006;71(1):30-39. doi: 10.1016/j.cardiores.2006.03.004.
Melnikov I, Kozlov S, Saburova O, et al. Monomeric C-reactive protein in atherosclerotic cardiovascular disease: advances and perspectives. Int J Mol Sci. 2023;24(3):2079. doi: 10.3390/ijms24032079.
McFadyen J, Kiefer J, Braig D, et al. Dissociation of C-reactive protein localizes and amplifies inflammation: evidence for a direct biological role of C-reactive protein and its conformational changes. Front Immunol. 2018;9:1351. doi: 10.3389/fimmu.2018.01351.
Kuta AE, Baum LL. C-reactive protein is produced by a small number of normal human peripheral blood lymphocytes. J Exp Med. 1986;164(1):321-326. doi: 10.1084/jem.164.1.321.
Fu Y, Wu Y, Liu E. C-reactive protein and cardiovascular disease: from animal studies to the clinic (Review). Exp Ther Med. 2020;2(2):1211-1219. doi: 10.3892/etm.2020.8840.
Zolotukhin I, Golovanova O, Efremova O, et al. Monocyte chemoattractant protein 1 plasma concentration in blood from varicose veins decreases under venoactive drug treatment. Int Angiol. 2022;41(6):457-463. doi: 10.23736/S0392-9590.22.04940-9.
Eschrich J, meyer R, Kuk H, et al. Varicose remodeling of veins is suppressed by 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. J Am Heart Assoc. 2016;5(2):e002405. doi: 10.1161/JAHA.115.002405.
Costa D, Andreucci M, Ielapi N, et al. Molecular determinants of chronic venous disease: a comprehensive review. Int J Mol Sci. 2023;24(3):1928. doi: 10.3390/ijms24031928.
Wadham C, Albanese N, Wang L, et al. High-density lipoproteins neutralize C-reactive protein proinflammatory activity. Circulation. 2004;109(17):2116-2122. doi: 10.1161/01.CIR.0000127419.45975.26.
Zhang H, Park Y, Wu J, et al. Role of TNF-α in vascular dysfunction. Clin Sci. 2009;116(3):219-230. doi: 10.1042/CS20080196.
Akhmedov A, Crucet M, Simic B, et al. TNFα induces endothelial dysfunction in rheumatoid arthritis via LOX-1 and arginase 2: reversal by monoclonal TNFα antibodies. Cardiovascular Research. 2022;118(1):254-266. doi: 10.1093/cvr/cvab005.
Virdis A, Colucci R, Bernardini N, et al. Microvascular endothelial dysfunction in human obesity: role of TNF-α. J Clin Endocrinol Metabol. 2019;104(2):341-348. doi: 10.1210/jc.2018-00512.
Virdis A, Duranti E, Rossi C, et al. Tumour necrosis factor-alpha participates on the endothelin-1/nitric oxide imbalance in small arteries from obese patients: role of perivascular adipose tissue. Eur Heart J. 2015;36(13):784-794. doi: 10.1093/eurheartj/ehu072.
Piao J, Hong H, Son Y. Substance P ameliorates tumor necrosis factor-alpha-induced endothelial cell dysfunction by regulating eNOS expression in vitro. Microcirculation. 2018;25(3):e12443. doi: 10.1111/micc.12443.
Deng X, Chu X, Wang P, et al. MicroRNA-29a-3p reduces TNFa-induced endothelial dysfunction by targeting tumor necrosis factor receptor 1. Mol Ther Nucleic Acids. 2019;18:903-915. doi: 10.1016/j.omtn.2019.10.014.
Castro-Ferreira R, Cardoso R, Leite-Moreira A, Mansilha A. The role of endothelial dysfunction and inflammation in chronic venous diseases. J Vasc Surg Vasc Lymphat Dis. 2018;6(4):552-553. https://doi.org/10.1016/j.jvsv.2018.05.013.
Hou T, Tieu B, Ray S, et al. Roles of IL-6-gp130 signaling in vascular inflammation. Curr Cardiol Rev. 2008;4(3):179-192. doi: 10.2174/157340308785160570.
Kang S, Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. 2021;53(7):1116-1123. doi: 10.1038/s12276-021-00649-0.
Su J, Luo M, Liang N, et al. Interleukin-6: a novel target for cardio-cerebrovascular diseases. Front Pharmacol. 2021;12:745061. doi: 10.3389/fphar.2021.745061.
Poredos P, Spirkoska A, Rucigaj T, et al. Do blood constituents in varicose veins differ from the systemic blood constituents? Eur J Vasc Endovasc Surg. 2015;50(2):250-256. doi: 10.1016/j.ejvs.2015.04.031.
Lattimer C, Kalodiki E, Geroulakos G et al. Are inflammatory biomarkers increased in varicose vein blood? Clin Appl Thromb Hemost. 2016;22(7):656-664. doi: 10.1177/1076029616645330.
Deng F, Wang S, Zhang L. Endothelial microparticles act as novel diagnostic and therapeutic biomarkers of circulatory hypoxia‐related diseases: a literature review. J Cell Mol Med. 2017;21(9):1698-1710. doi: 10.1111/jcmm.13125.
Nik I, Abdul Rahman R, Azlan M, et al. Endothelial microparticles as potential biomarkers in the assessment of endothelial dysfunction in hypercholesterolemia. Medicina (Kaunas). 2022;58(6):824. doi: 10.3390/medicina58060824.
Zhang J. Biomarkers of endothelial activation and dysfunction in cardiovascular diseases. Rev Cardiovasc Med. 2022;23(2):73. doi: 10.31083/j.rcm2302073.
Marei I, Chidiac O, Thomas B, et al. Angiogenic content of microparticles in patients with diabetes and coronary artery disease predicts networks of endothelial dysfunction. Cardiovasc Diabetol. 2022;21(1):17. doi: 10.1186/s12933-022-01449-0.
Han Y, Kim SY. Endothelial senescence in vascular diseases: current understanding and future opportunities in senotherapeutics. Exp Mol Med. 2023;55:1-12. doi: 10.1038/s12276-022-00906-w.
Wu CM, Zheng L, Wang Q, Hu YW. The emerging role of cell senescence in atherosclerosis. Clin Chem Lab Med. 2021;59(1):27-38. doi: 10.1515/cclm-2020-0601.
Schmidt TS, Alp NJ. Mechanisms for the role of tetrahydrobiopterin in endothelial function and vascular disease. Clin Sci (London). 2007;113(2):47-63. doi: 10.1042/CS20070108.
Fukumura D, Gohongi T, Kadambi A, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Nat Acad Sci USA. 2001;98(5):2604-2609. doi: 10.1073/pnas.041359198.
Wang L, Ge H, Peng L, Wang B. A meta-analysis of the relationship between VEGFR2 polymorphisms and atherosclerotic cardiovascular diseases. Clin Cardiol. 2019;42(10):860-865. doi: 10.1002/clc.23233.
Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis. 2006;9(4):225-230. doi: 10.1007/s10456-006-9055-8.
Nemtsova V, Bilovol O, Shalimova A. Vascular endothelial growth factor as a marker of endothelial dysfunction in poly- and comorbidity: focus on hypertension, type 2 diabetes mellitus and subclinical hypothyroidism. Arterial Hypertens. 2019;23(2):98-104. doi: 10.5603/AH.a2019.0006.
Patel S, Nilsson MB, Xiuning L, et al. Molecular mechanisms and future implications of VEGF/VEGFR in cancer therapy. Clin Cancer Res. 2023;29(1):30-39. doi: 10.1158/1078-0432.CCR-22-1366.
Li L, Wang H, Zhang J, et al. Effect of endothelial progenitor cell-derived extracellular vesicles on endothelial cell ferroptosis and atherosclerotic vascular endothelial injury. Cell Death Discov. 2021;7(1):235. doi: 10.1038/s41420-021-00610-0.
Mak A, Chan JKY. Endothelial function and endothelial progenitor cells in systemic lupus erythematosus. Nat Rev Rheumatol. 2022;18(5):286-300. doi: 10.1038/s41584-022-00770-y.
Heinisch PP, Bello C, Emmert MY, et al. Endothelial progenitor cells as biomarkers of cardiovascular pathologies: a narrative review. Cells. 2022;11(10):1678. doi: 10.3390/cells11101678.
Du F, Zhou J, Gong R, et al. Endothelial progenitor cells in atherosclerosis. Front Biosci. 2012;17(6):2327-2349. doi: 10.2741/4055.
Pelliccia F, Zimarino M, De Luca G, et al. Endothelial progenitor cells in coronary artery disease: from bench to bedside. Stem Cells Transl Med. 2022;11(5):451-460. doi: 10.1093/stcltm/szac010.
Altabas V, Bilos LSK. The role of endothelial progenitor cells in atherosclerosis and impact of anti-lipemic treatments on endothelial repair. Int J Mol Sci. 2022;23(5):2663. doi: 10.3390/ijms23052663.
Lenk K, Uhlemann M, Schuler G, Adams V. Role of endothelial progenitor cells in the beneficial effects of physical exercise on atherosclerosis and coronary artery disease. J Appl Physiol. 2021;111(1):321-328. doi: 10.1152/japplphysiol.01464.2010.
Akwii RG, Sajib MS, Zahra FR, Mikelis CM. Role of angiopoietin-2 in vascular physiology and pathophysiology. Cells. 2019;8(5):471. doi: 10.3390/cells8050471.
Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov. 2017;16:635-661. doi: 10.1038/nrd.2016.278.
Anisimov A, Fang S, Hemanthakumar KA, et al. The angiopoietin receptor Tie2 is atheroprotective in arterial endothelium. Nat Cardiovasc Res. 2023;2:307-321. doi: 10.1038/s44161-023-00224-y.
Sanchez FCL, Martinez GAC, Mendez-Garcia L, et al. Endoglin and other angiogenesis markers in recurrent varicose veins. J Pers Med. 2022;12(4):528. doi: 10.3389/fimmu.2018.01351.
Vischer UM. von Willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost. 2006;4(6):1186-1193. doi: 10.1111/j.1538-7836.2006.01949.x.
Gamal Y, Badawy A, Ali AM, et al. Endothelial dysfunction in children with newly diagnosed Graves’ disease. Eur J Pediatr. 2023;182(6):2793-2800. doi: 10.1007/s00431-023-04919-z.
Fan M, Wang X, Peng X, et al. Prognostic value of plasma von Willebrand factor levels in major adverse cardiovascular events: a systematic review and meta-analysis. BMC Cardiovasc Disord. 2020;20(1):72. doi: 10.1186/s12872-020-01375-7.
Peyvandi F, Garagiola, Baronciani L. Role of von Willebrand factor in the haemostasis. Blood Tranfus. 2011;9(Suppl 2):s3-s8. doi: 10.2450/2011.002S.
Springer TA. von Willebrand factor, Jedi knight of the bloodstream. Blood. 2014;124(9):1412-1425. doi: 10.1182/blood-2014-05-378638.
Schneider SW, Nuschele S, Wixforth A, et al. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci USA. 2007;104(19):7899-7903. https://doi.org/10.1073/pnas.0608422104.
Chauhan AK, Kisucka J, Lamb CB, et al. von Willebrand factor and factor VIII are independently required to form stable occlusive thrombi in injured veins. Blood. 2007;109(6):2424-2429. doi: 10.1182/blood-2006-06-028241.
Rezaie A, Giri H. Anticoagulant and signaling functions of antithrombin. J Thromb Haemost. 2020;18(12):3142-3153. https://doi.org/10.1111/jth.15052.
Song P, Xie J, Li W, et al. Effect of plasma thrombin-antithrombin complex on ischemic stroke: a systematic review and meta-analysis. Syst Rev. 2023;12(1):17. https://doi.org/10.1186/s13643-023-02174-9.
Gandrille S. Endothelial cell protein C receptor and the risk of venous thrombosis. Haematologica. 2008;93(6):812-816. https://doi.org/10.3324/haematol.13243.