
Introduction
Proteus syndrome (PS) is an extremely rare congenital multisystem disorder characterized by progressive, disproportionate, and asymmetric overgrowth of various tissues, including bone, skin, adipose tissue, and the central nervous system. The clinical presentation is highly variable, and no two individuals are affected in exactly the same way. To emphasize this phenotypic diversity, the term Proteus syndrome was proposed, referencing the ancient Greek sea god Proteus, who could alter his form at will.
Although the wide range of manifestations strongly indicated a genetic origin, the mode of inheritance remained unclear for decades. Early theories suggested a sporadic occurrence, but in 1993 it was hypothesized that somatic mosaicism could be the underlying mechanism. This hypothesis provided a plausible explanation for the segmental and patchy distribution of overgrowth seen in affected individuals, as well as the absence of vertical transmission.
A major breakthrough occurred in 2011, when Lindhurst et al. identified a mosaic activating mutation in the AKT serine/threonine kinase 1 (AKT1) gene (c.49G>A, p.E17K) in affected tissues of individuals with Proteus syndrome. This mutation leads to constitutive activation of the phosphoinositide 3-kinase (PI3K)/AKT1 signaling pathway, resulting in abnormal cell proliferation, survival, and growth. The identification of this postzygotic mutation confirmed the somatic mosaic nature of the disorder and provided a molecular basis for diagnostic confirmation in uncertain cases.
Due to its rarity, fewer than 200 cases have been reported worldwide, and the true prevalence of Proteus syndrome remains unknown. However, its clinical burden is profound, often involving life-threatening complications such as deep vein thrombosis, pulmonary embolism, and the development of benign or malignant tumors. In addition to the physical challenges, affected individuals frequently endure significant psychosocial suffering due to disfigurement and societal stigma.
Early recognition, multidisciplinary management, and regular follow-up are essential to mitigate medical risks and improve the quality of life for individuals living with this complex and enigmatic disorder.
Proteus syndrome (PS) is an ultrarare disease that manifests through combined damage to organs and systems [1-3]. The frequency of occurrence of PS is unknown, but is presumed to be 1:106–107 people. About 200 cases of PS are described in the world literature [3-5]. In Russia, according to open sources of information, fewer than 10 patients have been reported [6-12]. This syndrome was first described by Michael Cohen and Patricia Hayden (M.M. Cohen, P.W. Hayden) in 1979 [13]. To emphasize the variety of phenotypic features, the name “Proteus syndrome” was proposed – in honor of the ancient Greek god, capable of taking any form. A wide range of clinical manifestations in patients with PS indicated a genetic nature of the disease, but the type of inheritance remained unclear for a long time. In 1993, it was hypothesized that the syndrome is based on somatic mosaicism [14]. With the development of molecular genetic diagnostic methods, a search began for the gene responsible for the development of PS. Thus, X. Zhou et al., in 2000, suggested the involvement of the phosphatase and tensin homolog (PTEN) gene in the pathogenesis of PS [15]. Currently, it is believed that Proteus syndrome is based on postzygotic pathological variants of the AKT1 gene (14q32.33) [2]. The AKT1 gene is involved not only in the development of PS but also in several types of solid tumors – breast cancer, endometrial cancer, thyroid cancer, lung cancer, and malignant neoplasms of the genitourinary tract, etc. [16], which makes it a prospective target for the development of targeted therapy.
Case report
In 2010, an 18-year-old woman presented herself to the surgical team of a provincial hospital in Angola with an extremely unusual and emotionally charged request: elective amputation of her right forearm below the elbow. Her motivation was not driven by physical pain, functional limitation, or medical necessity, but by profound social ostracism and psychological distress. The patient had been the target of intense stigma and discrimination within her community, where some individuals unjustly accused her of being a manifestation of malevolent spiritual forces. This case highlights not only the clinical complexity of rare congenital disorders but also the severe moral and emotional suffering that patients may endure due to cultural beliefs and social exclusion.
We convey our profound gratitude to Professor Michele Garabedian and his team at National Institute for Medical Research in France (Unité 621), Saint-Vincent-de-Paul Hospital, 75014 Paris, which also includes highly experienced geneticists, who were kind enough to remotely examine the images (only the images) provided and confirm the diagnosis of Proteus syndrome.
The diagnosis in question was also confirmed by the co-author of this article, A. Olaru, an experienced specialist in oncology-traumatology at the Oncological Institute in Chișinău.
Family and perinatal history
The patient was born to healthy parents: the mother was 29 and the father 32 years old at the time of conception. The pregnancy was uneventful, aside from first-trimester hyperemesis gravidarum. She was delivered vaginally at term without complications. At birth, her weight was 3,500 grams, length 50 cm, and head circumference 33 cm. She had several siblings, all of whom were developmentally normal and healthy. There was no known family history of congenital anomalies or similar conditions.
This case highlights the tragic psychosocial consequences of congenital limb differences in certain cultural contexts and raises complex ethical considerations regarding bodily autonomy, stigma, and the role of medicine in addressing non-physical suffering.
Clinical findings
We observed marked asymmetry of the upper limbs, with significant elongation and disfigurement of the right forearm and hand. Two overlapping melanocytic nevi were noted at the left oral commissure, and a soft, mobile, non-tender retroauricular mass on the left side was identified, measuring approximately 7.0 × 5.5 cm and adherent to the auricle.
|
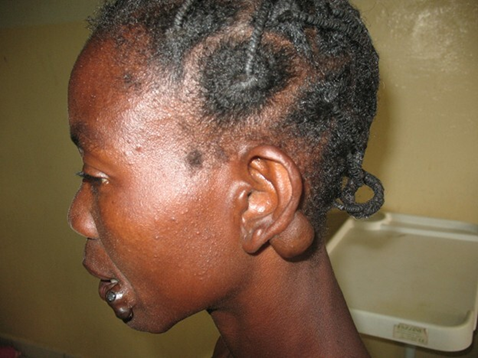
Fig. 1 Proteus syndrome Note: Two overlapping melanocytic nevi were noted at the left oral commissure, and a soft, mobile, non-tender retroauricular mass on the left side was identified, measuring approximately 7.0 × 5.5 cm and adherent to the auricle. |
|
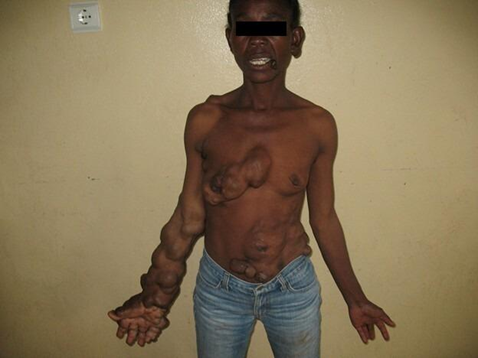
Fig. 2 Proteus syndrome Note: We observed marked asymmetry of the upper limbs, with significant elongation and disfigurement of the right forearm and hand. Soft tissue masses were also identified on the right side of the thorax, the anterior abdominal wall, and the left suprailiac region. |
|
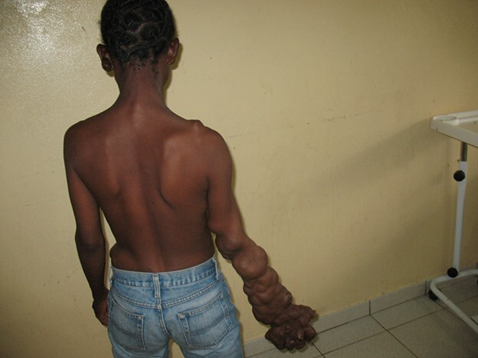
Fig. 3 Proteus syndrome Note: We observed marked asymmetry of the upper limbs, with significant elongation and disfigurement of the right forearm and hand. |
|
Fig. 4 Proteus syndrome Note: Significant elongation and disfigurement of the right forearm and hand. |

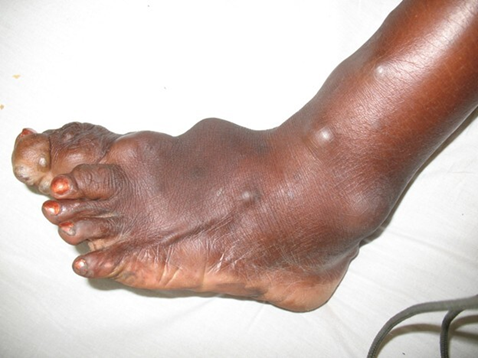
The right upper limb, particularly the arm and forearm, was longer by approximately 7.8 cm compared to the left, with evident vascular malformations along the forearm, digital hyperplasia, and deformation of the fingers. Soft tissue masses were also identified on the right side of the thorax, the anterior abdominal wall, and the left suprailiac region.
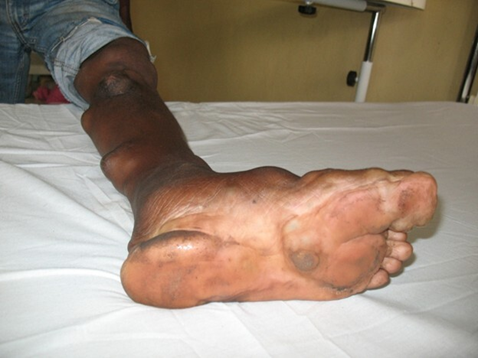
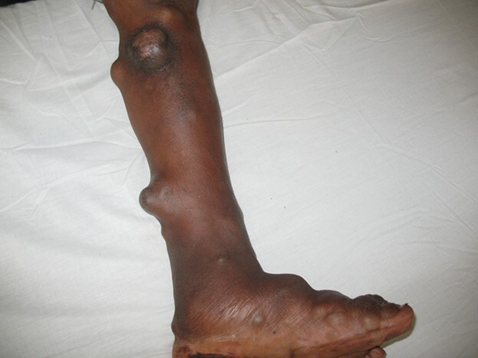
The left lower limb was slightly longer than the right by 2 cm, resulting in a tilted posture toward the right side, as visible in standing photographs. There was marked hypertrophy of the left calf (1.5 cm greater in circumference than the right), and the left foot exhibited soft tissue masses and hyperostoses on the surface of the calf, resembling the appearance of a tree trunk with multiple protruding branches. A pronounced plantar hyperkeratosis was also noted on the left foot.
|
Fig. 5 Proteus syndrome Note: The left lower limb was slightly longer than the right by 2 cm, resulting in a tilted posture toward the right side, as visible in standing photographs. There was marked hypertrophy of the left calf (1.5 cm greater in circumference than the right), and the left foot exhibited soft tissue masses and hyperostoses on the surface of the calf, resembling the appearance of a tree trunk with multiple protruding branches. |
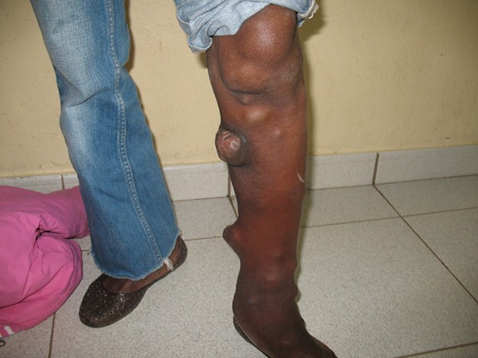
In addition to her depressive symptoms, the patient complained of pain and paresthesia in the right arm and forearm, as well as in the left calf. She reported sensory disturbances in the affected limbs, periodic fronto-occipital headaches, dysmenorrhea with associated pain (algodysmenorrhea), and increased fatigability. Her intellectual development appeared normal, and her behavior was appropriate for her age.
|
Fig. 6 Proteus syndrome Note: A pronounced plantar hyperkeratosis was also noted on the left foot. |
|
Fig. 7 Proteus syndrome Note: Digital hyperplasia and deformation of the fingers. |
|
Fig. 8 Proteus syndrome Note: There was marked hypertrophy of the left calf (1.5 cm greater in circumference than the right), and the left foot exhibited soft tissue masses and hyperostoses on the surface of the calf, resembling the appearance of a tree trunk with multiple protruding branches. |
No abnormalities were detected in other organ systems during the clinical examination. The diagnostic capacity of the hospital was extremely limited at the time, both in terms of laboratory tests and imaging modalities. The patient could not specify when the noted changes began but stated that she became aware of the differences around the age of 4–5, when she was able to compare herself to her siblings and other children. She reported that the deformities became more prominent between the ages of 10 and 13, after which they appeared to stabilize.
We would like to emphasize from the very beginning that we did not have the opportunity or the necessary resources to carry out genetic investigations in such cases.
Discussion
Proteus syndrome is characterized by a polymorphic phenotype. Clinical findings change over time, making this syndrome difficult to diagnose. It is an extremely rare disease, and its natural history is not yet fully understood [17]. The most widely accepted etiological hypothesis for this disease is genetic and is believed to represent the existence of somatic mosaicism. The disease is lethal in the non-mosaic state, and cases are mainly sporadic. Three adults reported in the literature as affected by the disease subsequently gave birth to normal children [18].
In several cases, asymmetry and hemihypertrophy have been observed at birth, although these features typically become more pronounced during postnatal development. Excessive growth of bones and soft tissues usually progresses throughout childhood and adolescence but tends to plateau after puberty. Despite localized overgrowth, overall height and pubertal growth spurts in patients with Proteus syndrome are generally within normal limits [19-22]. In this case, the basic physical parameters appear to be normal (waist: 1m 58cm, body mass: 52 kg, BMI: 20, head circumference: 54 cm).
The main long-term complications of Proteus syndrome include premature death and the development of unusual tumors. Premature mortality is most commonly attributed to deep vein thrombosis, which may lead to pulmonary embolism, a potentially fatal complication that requires vigilant surveillance and prophylactic measures [19-24]. Attention is drawn to lipomas, which are histologically benign tumors but can show invasive intra-abdominal or intrathoracic behavior.
Special attention should be given to lipomas, which, although histologically benign, may exhibit aggressive behavior, particularly when located intra-abdominally or intrathoracically. These lesions can infiltrate surrounding tissues, making surgical management challenging and increasing the risk of recurrence [19]. Due to the difficulty in identifying this syndrome, diagnostic criteria were established at the first National Conference on Proteus Syndrome in 1998. To make a diagnosis, all of these criteria should be present: mosaic distribution of lesions, progressive course, and sporadic occurrence of the disease (i.e., not familial) [19].
Various clinical manifestations may arise throughout the progression of Proteus syndrome. Among the most common features are hemihypertrophy, cranial hyperostosis, cerebriform nevi, pigmented nevi, subcutaneous tumors, vascular malformations, abnormal adipose tissue distribution, central nervous system involvement, and a range of ophthalmologic abnormalities. Less frequent findings include craniofacial anomalies, distinctive facial phenotypes, internal tumors, splenomegaly, and thymic hypertrophy [18, 19].
Hemihypertrophy typically develops during childhood and progresses until late adolescence. It may be partial, complete, or mixed, often resulting in limb dysfunction and gait disturbances. Treatment options include epiphysiodesis, arthrodesis, limb shortening or lengthening procedures, and various methods aimed at reducing asymmetry. Cerebriform nevi are connective tissue lesions characterized by excessive collagen deposition. They most commonly occur on the plantar surface of the foot and the palms of the hands. While not mandatory for diagnosis, their presence is considered almost pathognomonic for Proteus syndrome when identified.
Subcutaneous tumors (lipomas, hemangiomas, and lymphangiomas) develop variably in any part of the body. They can grow to infiltrate local tissues, making surgical resection difficult. Treatment options include resection, dissection, and liposuction; however, results are often unsatisfactory due to recurrence and the formation of hypertrophic scars.
Management strategies include excision, surgical dissection, and liposuction. However, treatment outcomes are often suboptimal due to the high risk of recurrence and the development of hypertrophic scars [20, 23]. Although the histopathological appearance of these tumors is benign, they can exhibit aggressive behavior, depending on their location, especially if they are intrathoracic or intra-abdominal [19]. Both increases and reductions in adipose tissue may occur simultaneously in different regions of the same patient, reflecting a disrupted regulation of adipose tissue homeostasis. A distinct pattern is often observed in association with lipomas and abnormal subcutaneous fat accumulation, and ectopic fat deposits may also be found between muscle layers, further highlighting the complex and dysregulated nature of adipose tissue in Proteus syndrome [20]. Vascular malformations may have a single component (e.g., capillary, lymphatic, or venous) or may be combined (e.g., capillary and venous, or capillary, venous, and lymphatic). They grow proportionally with the patient, never involute, and may expand [19]. Less common tumors associated with the syndrome include ovarian cystadenomas, meningiomas, testicular tumors, and parotid gland adenomas [18].
The present case is of interest as an ultra-rare condition, not encountered in daily practice by many practitioners worldwide. Treatment is largely palliative, and the complications that arise along the way shorten patients' lives. Moreover, the spectrum of clinical manifestations often overlaps with other genodermatoses, which complicates timely diagnosis. Based on the analysis of the literature, in most cases the diagnosis of PS was made based on clinical manifestations, as in our case, without performing the necessary genetic testing.
Management of these patients should be carried out by a multidisciplinary team, involving specialists in genetics, orthopedics, dermatology, neurology, radiology, and psychosocial care. Comprehensive clinical evaluations should be performed to identify the major manifestations of the syndrome, given its complex and heterogeneous presentation.
Long-term follow-up is essential due to the polymorphic nature of the disease, with the potential for progressive changes over time. Special caution is warranted when performing surgical procedures or during periods of prolonged immobilization, due to the increased risk of deep vein thrombosis and subsequent pulmonary embolism [18-24].
Surgical interventions in Proteus syndrome mainly involve the resection of large, highly vascularized tumors. Both conventional and ultrasound-assisted liposuction techniques have been tried, with the aim of reducing the volume of adipose tumors while preserving the surrounding vascular structures. However, the methods have proven insufficient, resulting in limited aesthetic and functional improvement.
In this case, the patient encountered significant difficulties and suffering due to isolation from major medical centers and the lack of access to specialized high-performance healthcare. Being located hundreds of kilometers from the capital, performing genetic testing, ultrasound examinations, and comprehensive biochemical laboratory analyzes was not feasible, although such evaluations are recommended by some authors [1]. Given the possible complications of this condition, patients with PS require lifelong surveillance and continuous risk prevention strategies, including [1]:
Annual examinations by a dermatologist, surgeon, orthopedist, pulmonologist, ophthalmologist, gynecologist/urologist, and oncologist (more frequently if clinically indicated);
Assessment of the blood coagulation system every six months;
Annual ultrasound examination of the veins of the lower extremities, particularly after the age of 25;
Annual pelvic ultrasound examination;
Annual chest X-ray.
Currently, the use of targeted therapy in the treatment of PS is not supported by clinical guidelines, but it may be considered in selected cases. This underscores the importance of timely diagnosis and molecular (DNA) testing [1].
NGS sequencing using a gene panel targeting the PI3K/AKT/mTOR signaling pathway, as emphasized by some authors from the Russian Federation [1], enables the differential diagnosis of phenotypically similar disorders and help determine the appropriate therapeutic strategy [1].
Consultation, ongoing supervision, and moral support from a qualified psychologist are extremely important and necessary for such patients. Without appropriate psychological support, individuals may develop severe depression, potentially leading to life-threatening consequences.
Conclusions
The description of the phenotypic characteristics of patients with Proteus syndrome is particularly important given the extreme rarity of this condition worldwide. It is necessary to increase the awareness and professionalism of clinicians regarding this disease in order to develop a dynamic follow-up plan, taking into account life-threatening complications (malignant tumors and risk of thromboembolism) and the moral suffering, sometimes unbearable, that patients encounter throughout their lives.
Today, the diagnosis of mosaic genodermatoses, including Proteus syndrome, is routinely performed in specialized laboratories. Molecular testing enables the detection of pathogenic variants in the AKT1 gene, even when present at low variant allele frequencies, thereby significantly increasing the rate of genetic confirmation. DNA-based diagnosis has become a critical tool not only for confirming clinical suspicion but also for guiding targeted therapies and conducting differential diagnosis with other disorders within the spectrum of segmental overgrowth syndromes.
Competing interests
None declared.
Authors’ contributions
ACV, NR, IR, AO, and CAV conceived the study. ACV, CAV, AO, and NR participated in the study design. ACV and IR helped to outline and draft the manuscript. ACV, CAV, and IR participated in the search and compilation of bibliographic sources. All authors critically reviewed the work and approved the final version of the manuscript.
Acknowledgements and funding
No external funding.
Patient consent
Obtained.
Provenance and peer review
Not commissioned, externally peer reviewed.
Authors’ ORCID IDs
Alexandru Voloc – https://orcid.org/0000-0001-8882-3164
Andrei Olaru – https://orcid.org/0009-0008-8298-7238
Ninel Revenco– https://orcid.org/0000-0002-5229-7841
Irina Rusu – https://orcid.org/ 0000-0002-2759-3171
Chiril Voloc – https://orcid.org/0000-0002-3428-9880
References
Belysheva TS, Zelenova EE, Semenova NA, Sharapova EV, Semenova VV, Sagoyan GB, et al. [Proteus syndrome: description of two clinical cases]. Voprosy Sovremennoi Pediatrii [Current Pediatrics]. 2024;23(5):343-349. Russian. https://doi.org/10.15690/vsp.v23i5.2797
Biesecker LG, Sapp JC. Proteus Syndrome. 2012 Aug 9 [updated 2023 May 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024 [cited 2025 May 12]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK99495/
Sapp JC, Hu L, Zhao J, et al. Quantifying survival in patients with Proteus syndrome. Genet Med. 2017;19(12):1376-1379. https://doi.org/10.1038/gim.2017.65
Sideris G, Nikolopoulos T, Sourla A, et al. Sinonasal neuroendocrine carcinoma in adult Proteus syndrome. Iran J Otorhinolaryngol. 2023;35(131):321-324. https://doi.org/10.22038/IJORL.2023.73128.3472
Khaladkar SM, Jhala NA, Krishnani KS, Durgi EC. Proteus syndrome: a rare congenital disorder. Cureus. 2024;16(5):e60072. https://doi.org/10.7759/cureus.60072
Semiachkina AN, Novikov PV, Voinova VIu, et al. [Proteus syndrome in children: diagnosis, treatment, prevention]. [Russ Bull Perinatol Pediatr]. 2007;52(1):45-9. Russian.
Il’ina EG, Erschova-Pavlova AA, Boyscha AS, et al. [A severe form of the Proteus syndrome in a newborn]. Med Genet. 2009;8(7):39-41. Russian.
Elizarova TV, Zriachkin NI, Khmilevskaia SA, et al. [Proteus syndrome in a child aged 14 years and 11 months]. Almanakh Clin Med. 2017;45(1):56-61. Russian. https://doi.org/10.18786/2072- 0505-2017-45-1-56-61.
Kriuchkova TA. Klinicheskii sluchai sindroma Proteia u rebenka rannego vozrasta [A clinical case of Proteus syndrome in an early age child]. Res Results Biomed. 2019;5(3):15-23. Russian. https://doi.org/10.18413/2658-6533-2019-5-3-0-3.
Nikolaeva IE, Raianova RR, Iakovleva LV. [A case of myocardial hypertrophy in Proteus syndrome]. Bashkortostan Med J. 2020;15(5):85-88. Russian.
Aleksandrov TI, Prokhorenko VM, Chornii SI, Simonova EN. Treatment of a patient with local gigantism of the upper limb: Proteus syndrome (case report). Mod Probl Sci Educ. 2022;(6). Russian. https://doi. org/10.17513/spno.32228.
Stozhkova IV, Pchelenok EV, Kosiakov SIa. Proteus syndrome in the practice of an otorhinolaryngologist: a clinical case. Russ Bull Otorhinolaryngol. 2020;85(2):45-48. Russian. https://doi. org/10.17116/otorino20208502145.
Cohen MM Jr, Hayden PW. A newly recognized hamartomatous syndrome. Birth Defects Orig Artic Ser. 1979;15(5B):291-6.
Cohen MM Jr. Proteus syndrome: clinical evidence for somatic mosaicism and selective review. Am J Med Genet. 1993;47(5):645-652. doi: 10.1002/ajmg.1320470514.
Zhou XP, Marsh DJ, Hampel H, et al. Germline and germline mosaic PTEN mutations associated with a Proteus-like syndrome of hemihypertrophy, lower limb asymmetry, arteriovenous malformations and lipomatosis. Hum Mol Genet. 2000;9(5):765-768. doi: 10.1093/hmg/9.5.765.
Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol. 2016;82(4):943-956. doi: 10.1111/ bcp.13021.
Cohen MM Jr. Further diagnostic thoughts about the Elephant Man. Am J Med Genet. 1988 Apr;29(4):777-82. doi: 10.1002/ajmg.1320290407.
Cohen MM Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. 2005;137C(1):38-52. doi: 10.1002/ajmg.c.30063.
Biesecker LG, Happle R, Mulliken JB, Weksberg R, Graham JM Jr, Viljoen DL, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84(5):389-95. doi: 10.1002/(SICI)1096-8628(19990611)84:5<389::AID-AJMG1>3.0.CO;2-O.
Clark RD, Donnai D, Rogers J, Cooper J, Baraitser M. Proteus syndrome: an expanded phenotype. Am J Med Genet. 1987;27(1):99-117. doi: 10.1002/ajmg.1320270111.
Cohen MM Jr, Turner JT, Biesecker LG. Proteus syndrome: misdiagnosis with PTEN mutations. Am J Med Genet A. 2003;122A(4):323-4. doi: 10.1002/ajmg.a.20474.
Gordon PL, Wilroy RS, Lasater OE, Cohen MM Jr. Neoplasms in Proteus syndrome. Am J Med Genet. 1995;57(1):74-8. doi: 10.1002/ajmg.1320570117.
Stricker S. Musculoskeletal manifestations of Proteus syndrome: report of two cases with literature review. J Pediatr Orthop. 1992 Sep-Oct;12(5):667-74.
Jamis-Dow CA, Turner J, Biesecker LG, Choyke PL. Radiologic manifestations of Proteus syndrome. Radiographics. 2004;24(4):1051-68. doi: 10.1148/rg.244035726.