
Introduction
Amyloidosis consists of a heterogeneous group of diseases in which proteins resulting from light chain amyloidosis (AL) immunoglobulin fragments are deposited in the extracellular space of organs and tissues, with the heart and kidneys being the most frequently involved. These amyloid deposits lead to the disorganization of the normal architecture of the affected organ or tissue, resulting in its progressive dysfunction [1, 2].
Amyloidosis is classified according to the biochemical nature of the fibrillar material derived from the precursor protein. Several heterogeneous types of amyloidosis are recognized, including primary amyloidosis, in which the protein precursor consists of immunoglobulin light chains, more commonly lambda (AL); secondary amyloidosis, where the precursor is serum protein A (AA); familial amyloidosis, with transthyretin (prealbumin) as the precursor protein; and the senile form (ATTR). Primary amyloidosis is part of the group of plasma cell dyscrasias characterized by clonal expansion, while the secondary form (AA) is a complication of chronic diseases such as rheumatic or inflammatory bowel diseases, tumors, and tuberculosis [3, 4].
From a clinical perspective, primary amyloidosis is differentiated into two subgroups: a) AL amyloidosis associated with multiple myeloma (about 20% of cases), which contains the Kappa light chain; and b) primary AL amyloidosis not associated with multiple myeloma (approximately 80% of cases), characterized by the lambda monoclonal chain [3].
Next, a unique clinical case for the Republic of Moldova is described involving a patient with primary hepatic amyloidosis not associated with multiple myeloma, presenting with acute liver failure and severe cholestasis that rapidly evolved into a fulminant state. Isolated liver damage in amyloidosis is a rare phenomenon, and this clinical case serves as an example of an atypical evolution of the disease.
Clinical case presentation
A 59-year-old woman, previously asymptomatic, was hospitalized in the Timofei Moșneaga Clinical Republican Hospital, Hepatology service, due to the onset of ascites, leg oedema, cholestatic syndrome, and severe liver insufficiency syndrome, which were detected incidentally in the biochemical profile conducted on an outpatient basis.
She had been considered ill since the beginning of December 2023, complaining of postprandial epigastric pain. An ambulatory upper digestive endoscopy demonstrated a healed duodenal ulcer, which was subsequently treated with proton pump inhibitors, leading to further remission of the symptoms. However, within a short period, her condition worsened due to the appearance of leg oedema and increased abdominal size. The patient had an uncomplicated medical history, with no history of alcohol consumption, smoking, or use of substances or medication.
Physical examination revealed a pale-brown complexion, clear sclera, and an enlarged abdomen due to ascitic fluid, along with massive leg oedema. Abdominal palpation demonstrated hepatomegaly, with the liver having an elastic consistency, and splenomegaly. The patient was hemodynamically stable.
Paraclinical investigations on admission revealed a minimal cytolytic syndrome and severe cholestatic syndrome, with elevated alkaline phosphatase (ALP) at 1245.90 U/L (norm: 30 - 120 U/L), gamma-glutamyl transpeptidase (GGT) at 599.40 U/L (norm: 9 - 39 U/L), and total cholesterol at 12.30 mmol/l (normal: 0 - 5.2mmol/l), while bilirubin levels remained normal at onset. Severe liver insufficiency with coagulation disorders was also observed: albumin at 20.80 g/L (norm: 35 - 53 g/L), total protein at 44.20 g/L (norm: 66 – 83 g/l), prothrombin index at 39.10%, and International Normalized Ratio (INR) at 1.86.
Liver damage from viral and autoimmune causes was ruled out by testing for viral hepatitis markers, dsDNA antibodies, antimitochondrial antibodies (AMA-M2), antinuclear antibodies, and anti-PR3 antibodies (ANCA), all of which were within reference values. The cytology of the ascitic fluid was unremarkable, with no atypical cells identified. Urinary protein electrophoresis with immunofixation for Bence Jones proteins was negative.
Abdominal ultrasound confirmed the presence of ascites and hepatosplenomegaly. The mechanical etiology of cholestasis was assessed via magnetic resonance cholangiopancreatography (MRCP), which revealed only significant biliary sludge. Contrast-enhanced computed tomography confirmed these findings. An outpatient abdominal MRI with contrast demonstrated significant diffuse ascites and hepatosplenomegaly, with no evidence of portal hypertension. However, a dysplastic Li-RADS III nodule was identified in segment 4 (S4) without signs of malignancy.
Given the patient’s deteriorating condition and inconclusive clinical and paraclinical findings, a diagnostic laparoscopy with liver biopsy was performed. Macroscopic examination revealed an enlarged liver with sharp edges, a brownish-violet color, and a firm texture. In S4b, a tumor mass approximately 2 cm in diameter was observed.
Histological examination of the liver biopsy sample revealed altered trabecular architecture, with hepatocellular oedema and swelling accompanied by protein degeneration. The hepatocytes exhibited hyperchromatic nuclei of uneven size, a slight increase in the nucleocytoplasmic ratio, occasional intranuclear inclusions, and mildly prominent nucleoli. Diffuse capillarization of the sinusoids was identified using Masson’s trichrome stain, while the reticular architecture remained preserved as shown by Reticulin staining (Fig. 1a, b.).
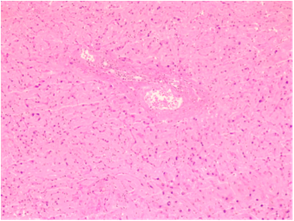 | 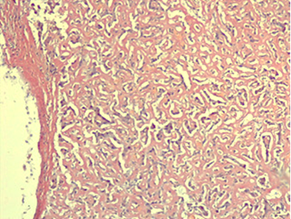 |
Fig. 1 AL hepatic amyloidosis. a. Perisinusoidal pattern of amyloid deposit (staining: hematoxylin-eosin). b. Disruption of liver architecture marked by diffuse extracellular and vascular wall accumulations of amorphous, acellular eosinophilic material, causing noticeable atrophy of focal hepatocytes (staining: Congo red). | |
The positive Congo Red staining test was validated by a control test under polarizing microscopy, which confirmed amyloid deposits in the vascular wall, capsule, and perisinusoidal areas. Under polarizing microscopy, these deposits exhibited characteristic apple-green birefringence (Fig. 2 a, b), creating a microscopic picture indicative of hepatic amyloidosis.
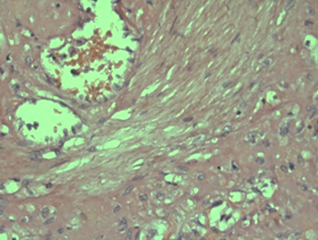 | 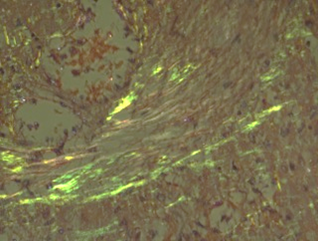 |
Fig. 2 AL hepatic amyloidosis. a. Vessel with amyloid deposits observed under optical microscopy (staining: Congo red); b. Amyloid deposits in the vessel under polarizing microscopy. | |
Given the frequent association of primary amyloidosis with multiple myeloma and Waldenstrom's macroglobulinemia, the patient underwent a percutaneous bone marrow biopsy. The myelogram revealed a hypocellular bone marrow, with the granulocytic series constituting 69.0%, segmented neutrophils accounting for 57%, and multiple macrophages observed. Megakaryocytes were within normal limits, and platelets showed a slight increase.
In the 3-7 weeks following hospitalization, the patient's condition deteriorated, leading to the development of hepatorenal syndrome, with creatinine levels reaching 446 µmol/L (norm: 44 – 115 µmol/L), and urea at 36.2 mmol/L (norm: 2.8 – 7.2 mmol/L), accompanied by oliguria. Liver failure and cholestasis continued to progress. The patient subsequently developed septicemia, with blood cultures positive for Escherichia coli, and was diagnosed with mycotic esophagitis during upper digestive endoscopy. Additionally, bilateral basal pneumonia and left-sided pleurisy emerged as complications over time.
Treatment included corticosteroids, substitution therapy, antibiotic therapy, antifungal treatment, and supportive care. Despite the efforts of a multidisciplinary team consisting of a gastroenterologist, hematologist, oncologist, and radiologist, the patient passed away two months after admission. Liver transplantation and chemotherapy were not recommended due to concurrent hospital-acquired infections and multi-organ failure.
Discussion
A case of a patient with acute liver failure and severe cholestasis as the primary manifestations of AL amyloidosis is reported. The patient presented with jaundice, hepatomegaly, leg oedema, ascites, preserved renal function, hepatic failure, and severe cholestatic syndrome, characterized by alkaline phosphatase (ALP) and gamma-glutamyl transpeptidase (GGT) levels exceeding 20 times the upper limit of normal.
Involvement of the liver in primary amyloidosis is reported in only 3.9% of cases, frequently being asymptomatic at onset [5]. The clinical picture varies, with pronounced physical asthenia, peripheral oedema, and headache being the most common manifestations. Jaundice is present in about 5% of cases, typically associated with moderate to severe cholestatic syndrome [2].
In a retrospective clinical study evaluating 98 patients with hepatic amyloidosis, 88% of them presented with proteinuria, 86% with increased ALP, 81% with hepatomegaly, and 62% with hyposplenism on peripheral blood smear. Similarly, the patient had hepatomegaly and elevated ALP but no other associated features [6]. In another study, Nakano Y. et al. reported that 80% of patients with hepatic amyloidosis exhibited hypercholesterolemia. As demonstrated in this clinical case, the patient had a total cholesterol level of 12.30 mmol/L. Therefore, physicians should consider amyloidosis as a potential cause of secondary hypercholesterolemia, especially after ruling out nephrotic syndrome [7].
Imaging findings are nonspecific. On contrast-enhanced computed tomography, liver areas infiltrated by amyloid typically appear as hypoattenuating focal lesions. In this clinical case, however, the liver parenchyma presented multiple lesions with increased contrast up to +80 HU, more suggestive of dysplastic regenerative nodules, well-known indicators of liver cirrhosis [8].
A positive bone marrow biopsy for amyloid is reported in 95% of cases in patients with primary hepatic involvement. However, in this case, the bone marrow biopsy was negative for amyloid [9].
Primary hepatic amyloidosis presents with a heterogenous clinical profile that can mimic other hematological diseases, such as lymphoma, or various liver diseases, including drug-induced liver injury and Budd-Chiari syndrome. Diagnosis is confirmed by histopathological examination where the liver parenchyma will test positive for Congo red and show apple-green birefringence under polarized light microscopy. In this case, spectroscopic analysis could not be performed due to technical issues in identifying the type of amyloid protein [9, 10]. Early diagnosis of amyloidosis is crucial for effective therapy, as it allows for better chemotherapy tolerability [11].
Primary amyloidosis has a poor prognosis, with an average survival rate of up to 9 months despite chemotherapy treatment [12]. When patients develop jaundice with bilirubin levels exceeding 34 umol/L, the prognosis worsens considerably, with median survival ranging from one to 3.3 months [13, 14]. In this case, despite high-dose corticosteroid treatment, replacement therapy, and palliative care, the patient survived for 1.5 months following the progression of cholestatic syndrome.
Data on liver transplantation in patients with primary amyloidosis is limited. A retrospective study reported that 9 patients who underwent transplantation had 1- and 5-year survival rates of 33% and 22%, respectively [15].
Conclusions
Due to nonspecific clinical and imaging findings, early diagnosis of primary hepatic amyloidosis is challenging and requires a multidisciplinary approach. Severe cholestasis and liver failure are indicators of poor prognosis and limited treatment response. A meticulous differential diagnosis, including infiltrative diseases such as amyloidosis, is essential when a patient presents with cholestasis and deteriorating liver function. Future studies should focus on developing specific therapies for primary amyloidosis to improve survival rates in these patients.
Competing interests
None declared.
Authors’ contribution
ET conceived the study and contributed to the study design. AMB also conceived the study, participated in the study design, and helped draft the manuscript. COS assisted in drafting the manuscript and made a substantial contribution to data acquisition. RP performed immunofluorescence tests and participated in staining. EB critically reviewed the article for significant intellectual content. KB made a substantial contribution to data acquisition. All authors critically reviewed the work and approved the final version of the manuscript.
Informed consent for publication
Obtained.
Funding
None.
Authors’ ORCID IDs
Eugen Tcaciuc – https://orcid.org/0000-0002-2362-1788
Elina Berliba – https://orcid.org/0000-0002-3142-5218
Kalina Bugor – https://orcid.org/0009-0000-4452-8593
Ruslan Pretula – https://orcid.org/0000-0002-4785-3647
Cătălina Olaru-Stăvilă – https://orcid.org/0009-0005-3524-427X
Ana Maria Bădărău – https://orcid.org/0009-0000-3105-1685
References
Sonthalia N, Jain S, Pawar S, Zanwar V, Surude R, Rathi PM. Primary hepatic amyloidosis: a case report and review of literature. World J Hepatol. 2016 Feb 28;8(6):340-4. doi: 10.4254/wjh.v8.i6.340.
Dias T, Ferreira D, Moreira H, Nascimento T, Santos A, Carvalho A. A Case of severe cholestasis due to hepatic AL amyloidosis. GE Port J Gastroenterol. 2019 Oct;26(6):425-429. doi: 10.1159/000496185.
Gavilán JC, Bermúdez FJ, Márquez A, Sánchez-Carrillo JJ, González-Santos P. Amiloidosis hepática como causa de colestasis severa intrahepática [Hepatic amyloidosis as cause of severe intrahepatic cholestasis]. An Med Interna. 2003 Jan;20(1):25-7. Spanish.
Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJ, Westermark P. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016 Dec;23(4):209-213. doi: 10.1080/13506129.2016.1257986
Brouwers S, Heimgartner R, Laptseva N, Aguzzi A, Ehl NF, Fehr T, et al. Historic characteristics and mortality of patients in the Swiss Amyloidosis Registry. Swiss Med Wkly. 2024 Feb 15;154:3485. doi: 10.57187/s.3485.
Park MA, Mueller PS, Kyle RA, Larson DR, Plevak MF, Gertz MA. Primary (AL) hepatic amyloidosis: clinical features and natural history in 98 patients. Medicine (Baltimore). 2003 Sep;82(5):291-8. doi: 10.1097/01.md.0000091183.93122.c7.
Nakano Y, Kawamoto R, Ito E, Matukawa K. A case of cholestatic liver involvement secondary to amyloid light chain amyloidosis with new-onset hypercholesterolemia and elevated gamma-glutamyltransferase level. Cureus. 2023 Aug 23;15(8):e44001. doi: 10.7759/cureus.44001.
Matsuda S, Motosugi U, Kato R, Muraoka M, Suzuki Y, Sato M, et al. Hepatic amyloidosis with an extremely high stiffness value on magnetic resonance elastography. Magn Reson Med Sci. 2016 Jul 11;15(3):251-2. doi: 10.2463/mrms.ci.2015-0133.
Son RC, Chang JC, Choi JH. Primary hepatic amyloidosis: report of an unusual case presenting as a mass. Korean J Radiol. 2011;12(3):382-385. doi: 10.3348/kjr.2011.12.3.382.
Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJ, Westermark P. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016 Dec;23(4):209-213. doi: 10.1080/13506129.2016.1257986.
Gertz MA. Immunoglobulin light chain amyloidosis: 2020 update on diagnosis, prognosis, and treatment. Am J Hematol. 2020 Jul;95(7):848-860. doi: 10.1002/ajh.25819.
Gertz MA, Kyle RA. Hepatic amyloidosis (primary [AL], immunoglobulin light chain): The natural history in 80 patients. Am J Med. 1988;85:73–80.
Ubiña Aznar E, Fernández Moreno N, Rivera Irigoín R, Moreno Mejías P, Fernández Pérez F, Vera Rivero F, et al. Massive hepatic amyloidosis with fatal hepatic failure. Rev Esp Enferm Dig. 2006 Jul;98(7):551-2. English, Spanish. doi: 10.4321/s1130-01082006000700009.
Econimo L, Jeannin G, Biasi L, Mazzola G, Mendeni M, Presteni K, Gregorini G. Liver involvement with rapidly progressive course in light chain (AL) amyloidosis: distinguishing features at presentation. Experience of a single center. Amyloid. 2011 Jun;18 Suppl 1:109-11. doi: 10.3109/13506129.2011.574354040.
Zhang X, Tang F, Gao YY, Song DZ, Liang J. Hepatomegaly and jaundice as the presenting symptoms of systemic light-chain amyloidosis: a case report. World J Gastrointest Oncol. 2024 Feb 15;16(2):550-556. doi: 10.4251/wjgo.v16.i2.550.