
Introduction
Inborn errors of metabolism (IEM)are a phenotypically and genetically heterogeneous group of disorders, caused by mutations in specific genes that encode enzymes, membrane transporters, and other functional proteins that produce a block on a metabolic pathway, leading to the toxic accumulation of substrate and / or a deficiency of an important biological product [1]. More than 1000 IEM have been described [2]. The cumulative prevalence of IEM in different populations is around 1:500–800 newborns [3-5], despite the fact that some of these disorders are extremely rare when taken individually [6]. Most of IEM are inherited in an autosomal recessive way. A minority of them is transmitted as autosomal dominant, X-linked or by mitochondrial DNA only of maternal origin, as in subsets of respiratory chain disorders [7, 8].
Most of the ”intoxication type” of the IEM are potentially treatable and appropriate diagnosis and therapeutic measures must be initiated as soon as possible to avoid long-term damage [9]. The diagnosis of IEMs is based on evaluation of clinical manifestations of the patient and metabolic workup including specialized analyses as blood and urine amino acids, blood spot acylcarnitine profile, urine organic acids, and specific analytical tests, in order to identify abnormally high and / or low levels of metabolites caused by the enzymatic blockage of a metabolic pathways [10]. Quantitative amino acids analysis is an important tool for the diagnosis of “intoxication type” of IEM as: aminoacidopathies, urea cycle disorders, some organic acidurias as well as it could offers some clues for mitochondrial disorders.
In this context the aim of the study was defined as evaluation of importance of plasma amino acid profile in the diagnosis of IEM. As the diagnosis is performed, specific biomarkers are followed to monitor clinical management of the patient.
Material and methods
This analytical prospective study was done within the project «Genomic medicine and metabolomics research in the service of prophylaxis of genetic diseases for healthy generations in the Republic of Moldova» (Acronym: SCREENGEN, Cipher: 20.80009.8007.22) in Institute of Mother and Child from Chisinau, Republic of Moldova. The research protocol was approved by the Ethics Research Committee of USMF “Nicolae Testemitanu”, from the Republic of Moldova (verbal process no. 2 from 15.12.2020, signed by the president of the committee – Professor Vovc Victor).
Amino acids analysis was performed in 15 children aged from 0 to 13 years old, which parents signed inform consent after receiving all necessary information. The subjects were chosen through medical genetic consulting based on clinical manifestations specific for “intoxication type” of IEM. In the study there was included any newborn or child without particularities of pregnancy and birth in medical history, but who presents a sudden and progressive deterioration of health associated with: acid-base imbalance, hypoglycemia, lethargy, coma, vomiting, hyperammonemia, developmental delays, unexplained liver function disorder, cardiac signs (arrhythmias, cardiomyopathies, cardiomegaly), unexplained neurological symptoms (acute encephalitis, intellectual disabilities, seizures), reduced consciousness, especially after episodes of vomiting, fever or hunger [11-13]. Children with a confirmed metabolic disease, which do not interfere with amino acid metabolism, were excluded from the study, the same for the patients who did not sign the informed consent.
A complex metabolic workup have been performed in the patients with unclear diagnosis including first line investigations – routine tests realized in the hospital units and second line investigations that cover more complex analysis, realized in specialized laboratory, as plasma amino acids by high performance liquid chromatography (HPLC) at the Institute of Mother and Child, Chisinau, Moldova, metabolic extended screening from dried blood spots (DBS) by liquid chromatography-tandem mass spectrometry (LC-MS/MS) at CytoGenomic Medical Laboratory, Bucharest, Romania and urine organic acids by Nuclear Magnetic Resonance (NMR) Spectroscopy, at „Petru Poni” Institute of Macromolecular Chemistry of Romanian Academy, Iasi, Romania.
The selected patients were subjected to biological fluid collection, as blood and urine. Peripheral blood was drawn by venous puncture on fasting and collected into 5 mL K3EDTA-coated tubes. Then the blood was centrifuged (4500 rpm for 10 min), and plasma was transferred to 1.5 mL Eppendorf tubes and stored at - 80°C until chromatographic analysis were done.
Before analysis fat-soluble components and proteins have been removed from plasma, then the obtained mix was filtered through syringe filter, 0,45 µm membrane pore sizes in a 2 ml volume vial and introduced in the amino acid analyzer. Amino acids were profiled by High Performance Liquid Chromatography (HPLC, Shimadzu) with chromatographic column Shim-pack Amino-Na, 100 mmL × 6,0 mmI.D., 5 μm, post-column derivatization with O-pthalaldehyde (OPA) and fluorescence detector. The analytical procedure used in this research for amino acids quantification was performed as described in Shimadzu manual „Amino Acid Analysis System for high-sensitivity measurement of amino acids in foods and biological samples”. The chromatographic method was calibrated using „Amino acid standards, physiological, analytical standard, acidic, neutrals, and basics”, Merck, Germany. Norvaline was used as internal standard.
Samples of urine were collected usually overnight from patients aged 3-13 years in special collection tubes and stored in the fridge (+ 40C) for at most 8 hours or in the freezer at - 200C for a longer period of storage. In contrast, newborns and infants sample required special collector bags that allow acquisition of urine randomly during the day.
Results
During the study, 15 patients have been selected for analysis according to inclusion and exclusion criteria, described in table below. (Table 1).
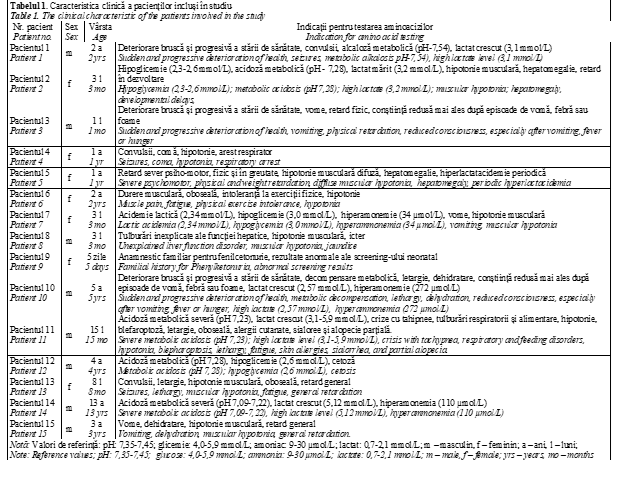
The first line metabolic investigations including acid-base balance, glucose, lactate and ammonia level have been done during patients hospitalization in all cases. In 33,3% (n = 5) there has been observed acid-base imbalance in order of severe acidosis, in 20% (n = 3) – hypoglycemia, in 46,6% (n = 7) – high lactate level and 20% (n = 3) of the children had high ammonia level. Metabolic extended screening has been performed in 13 patients (86,66%). The described patients were subjected to chromatographic analysis by HPLC and the obtained chromatograms were suggestive for an inborn error of metabolism in 4 cases (26,6%).
Abnormal results on plasma amino acids have been seen in patient number 4, a girl of 2 months old, whose main clinical conditions were seizures from birth, precomatous state and respiratory arrest. From the third day of life, the child started to develop intractable seizures then she went into a coma for a month. On electroencephalography there was observed excessive discontinuity of cerebral activity in the form of burst-suppression pattern and separated in the frontal region with an emphasis on the left isolated acute hypervolted sharp waves of epileptiform character. Computed tomography scan showed cerebral mixed hydrocephalus.
Based on clinical manifestations and evolution, an IEM has been suspected. In this context, body fluids samples (DBS, plasma, urine, cerebrospinal fluid (CSF)) have been collected. Amino acids analysis in CSF, plasma and urine has been performed by HPLC (Fig. 1) and ion-exchange liquid chromatography (Institute of Physiology and Sanocreatology, Chisinau, Moldova).
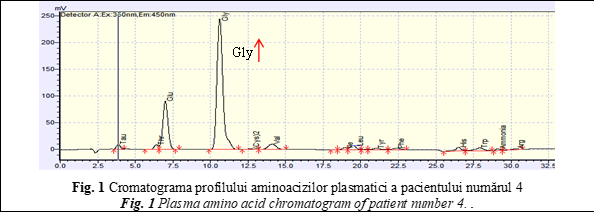
The obtained amino acid profile showed high level of glycine (Gly) in plasma (544 μmol/L; reference value 70,72 – 256,36 μmol/L), being suggestive for disorder of Gly metabolism – Nonketotic hyperglycinemia (NKH). It is known that the biochemical hallmark for this disease is the elevated glycine CSF / plasma ratio, so the amino acids analysis was performed in CSF. Gly level was also high, exceeding 10 fold above the normal ranges (reference value: <20 μmol/L) and the Gly CSF / plasma ratio of 0,147 (normal<0.02) was evident for NKH [17, 18]. Additionally, the data from newborn extended screening have been fortified the diagnosis (glycine values – 1085,9 μmol/L (reference value: < 650,0 μM/L) as well as the result from NMR spectroscopy, identifying high amount of Gly in the urine. The diagnosis was confirmed by two pathological mutations found in homozygous state in the GLDC gene during molecular investigations associated with Glycine encephalopathy [OMIM 605899].Consequently, the treatment has been individualized according to the biochemical diagnosis. So, the patient therapy included a low glycine diet and the sodium benzoate 200 mg/kg/day administration.
Another case with abnormal amino acid profile was the patient number 9, a 5 days old girl who had a familial history for Phenylketonuria (PKU), but did not perform the prenatal diagnosis. The older brother has been diagnosed with PKU by Moldavian National Screening Program 4 years ago. In this context dried blood spots sample has been collected from the newborn girl in the third day of life, for early diagnosis of PKU. Her results were abnormal (Phenylalanine >14 mg/dl; reference values <3 mg/dl), being suggestive for disorders of phenylalanine (Phe) metabolism. Plasma amino acids analysis has been done to confirm abnormal newborn screening results and quantify the exact concentration of plasma Phe and tyrosine amino acid (Tyr), in order to initiate and personalize the diet-therapy [14, 15]. Amino acid profile showed a high peak at retention time – 22,5 minutes, corresponding with amino acid Phe (Fig. 2).
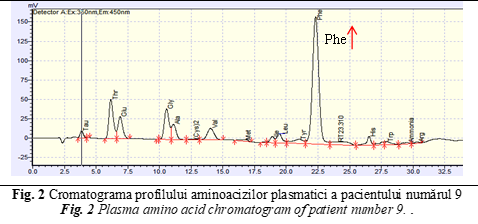
Analyzing the obtained data, extremely high concentrations of Phe (1568 µmol/L; reference values 26.52-221 µmol/L) have been detected, that confirmed disorder of phenylalanine metabolism. Additionally, the tyrosine (Tyr) has been identified in normal concentration (92,4 µmol/L; reference values 26,52-123,76 µmol/L), but the Phe / Tyr ratio was around 17 and it was specific for classical form of PKU [17]. Genetic analysis of PAH gene, performed by Sanger sequencing confirmed the PKU disease.
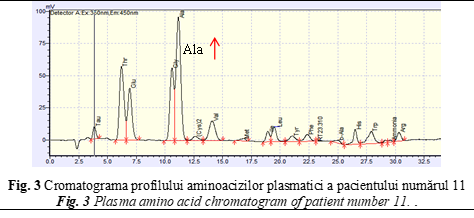
The next reported child with abnormal level of amino acid profile was patient number 11, a 15 months old boy. The onset of clinical presentations appeared at 15 months of life, manifested by severe metabolic acidosis crisis with tachypnea, respiratory and feeding disorders, hypotonia, blepharoptosis, lethargy, fatigue, skin allergies, sialorrhea, and partial alopecia (Table 1). Considering all clinical manifestations, the child was suspected for an IEM. Plasma amino acids analysis has been performed by HPLC in Institute of Mother and Child and State University of Medicine and Pharmacy ”Nicolae Testemitanu”. Both HPLC analysis of plasma amino acids revealed high concentration of alanine (Ala – 572 µmol/L, reference values: 79.56 – 424.32 µmol/L), along with the value of the ratio Ala/Lys of 6,8 indicated the mitochondrial involvement (Fig. 3). Extended newborn screening by LC-MS/MS of DBS samples showed high concentration of propionil-L-carnitina (C3 = 3,504 μM/L, (reference values < 2,7 μM/L), hydroxy isovaleric-L-carnitina (C5OH = 2,317 μM/L, (reference values < 0,65 μM/L), and high ratio of C5OH/C0 – 0,076 (reference values < 0,035), and C5OH/C8 – 75,274 (reference values < 13). Additionally, by performing organic acids analysis in urine, high levels of 3-hydroxyisovaleric acid, 3-hydroxypropionic acid and 2-methylcitric acid has been identified in the presence of pyruvic and lactic acidemia. No ketone bodies have been identified in the urine.
Based on clinical manifestations and laboratory data, there has been observed a strong suspicion for a disorder of biotin metabolism – Multiple CoA carboxylase deficiency (MCD), well treatable by oral biotin supplementation. In this case, the patient received 20-40 mg biotin per day, without any diet. The patient was well responsive to the treatment, the following urine monitoring revealed disappearance of toxic metabolites and the clinical state has been improved. The patient is continuing the biotin supplementation and is maintained in a good metabolic and health condition.
Patient number 14 was the last with abnormal amino acid profile in the analyzed cohort. At 13 years old, he was hospitalized with severe metabolic acidosis (Table 1), possible triggered by Covid-19 infection and excessive protein intake during winter holidays. His symptoms (myoglobinuria / rhabdomyolysis, ketosis, hyperammonemia, Reye's - like syndrome (fulminant liver failure and central nervous system damage)) in the context of personal history (myopathy, fatigue, physical exercise intolerance, frequent vomiting started from 7 years old) made him suspected for an IEM. In order to elucidate the cause of these abnormalities, the metabolic workup has been performed. The HPLC analysis of plasma showed increased concentration of Ala (575 µmol/L; reference values: 192-508 µmol/L). Acylcarnitine profile results revealed slightly increased concentration of 3-hydroxyisovaleryl-/2-methyl-3-hydroxybutyryl-carnitine (C5OH-Carnitine - 0,840 μM/L, reference values < 0,65 μM/L) and high ratio of C5OH/C0 (0,037 μM/L, reference values < 0,035 μM/L). Urine organic acids revealed high concentration of lactic acid, 3-hydroxibutiric acid, 2-hydroxibutiric acid, 3-hydroxivalerianic acid, 2-hydroxivalerianic acid and 3-methylglutaconic acid. Combining all the data a possible presence of a disorder with mitochondrial involvement was suspected. Whole exome sequencing did not reveal any pathological mutation, so the mitochondrial DNA sequence was recommended. He responded well to biotin treatment with normalization of urine profile and clinical data as well.
Discussions
Despite to the fact that IEM may present at various ages, the disorder of amino acids pathway is present from birth and may be manifested the first days of life as deterioration after normal birth and delivery or later as nonspecific chronic manifestations [11]. Acute metabolic decompensation in the neonatal period may also present severe acidosis (blood pH below 7,35), alkalosis (blood pH above 7,45), hypoglycemia, high ammonia and lactate levels [6, 19] due to accumulation of toxic metabolites in the body. Advanced laboratory tests have been implemented in order to detect specific biochemical biomarkers associated with particular IEM. Among these tests amino acid analysis plays a crucial role and should be done in every patient with a suspected IEM.
Amino acids quantification is a perfect tool for diagnosis of aminoacidopathies and can offer complementary data for other types of IEM. The interpretation of amino acids profile is based not only on the abnormal level of a single amino acid, but rather involves other specific biomarkers including diagnostic ratios (Table 2) and correlation to the patient’s clinical history. Biomarkers serve as measurable indicators of normal biological or pathological processes; they can predict, diagnose, monitor and assess the severity of a disease. For example, detection of high level of plasma Phe is associated with PKU [OMIM 261600], an inherited disorder of phenylalanine metabolism, caused by mutations in PAH gene (around 98% cases) and in rare cases by mutations in genes implicated in biosynthesis of tetrahydrobiopterin [19]. Along with elevated Phe (concentration > 120 μmol/L), an elevated Phe / Tyr ratio (> 3) is an important biomarker for the diagnosis of PKU, that can be used to differentiate between PKU and hyperphenylalaninemia (HPA) [20]. Additional biomarkers for PKU are obtained by performing urine organic acid analysis; patients with PKU will show elevations in phenylpyruvic, phenylacetic, phenyllactic, 2-hydroxyphenylacetic, and N-phenylacetylglutamic acids (Table 2) [3]. Individuals with PKU have Phe and Tyr routinely monitored, to ensure optimal dietary control of the disease. The importance of biomarkers from amino acids spectrum can be seen on example of Nonketotic hyperglycinemia, also known as glycine encephalopathy. As NKH is an aminoacidopathie, the diagnosis relies on amino acid analysis, which should reveal an elevation of glycine in body fluids (plasma, urine, and CSF). However, plasma glycine level may not be significantly increased in all cases due to diurnal variation and differences in disease severity, but glycine is always elevated in CSF. CSF collection is recomended in any case when the intractable seisures are present in the patient from the birth, for the following metabolic investigations. Therefore, the elevated CSF / plasma ratio, usually greater than 0.08 in classical NKH, and greater than 0.04 in atypical NKH can be used for diagnosis of NKH [21].

In case of alanine, high concentration of this amino acid is not directly associated with pathology but acts as secondary biomarker in different disorders. It is interesting that elevated Ala levels (> 450 µmol/L) are suggestive for a mitochondrial dysfunction, but it is nonspecific and not diagnostic marker [21].
In this context interpretation of results from metabolic laboratory data must be complex, with regard to all available biomarkers. Likewise, prior to the analysis and results interpretation, pre-analytical variables such as a dietary status and medication treatments should be taken into account in order to avoid false positive results [22]. For example, certain drugs, including some antidepressants and antiepileptics, can produce an elevation in glycine, which could be misdiagnosed as nonketotic hyperglycinemia [3]. A diet rich in proteins leads to increased excretion of β alanine and 1-methylhistidine, thus blood collection intended for amino acids analysis is recommended after overnight fasting. By contrast, a decreased protein intake or increased catabolism due to vomiting or fasting can result in elevations of branched chain amino acids (BCAA), potentially mimicking Maple Syrup Urine Disease (MSUD) [21]. The body fluids collection and storage may influence the amino acids level as well. As example, a hemolyzed sample may lead to the decrease of arginine with simultaneous increase of ornithine due to red blood cells arginase activity, and an increase in taurine that released from leukocytes and platelets. Overall, during a prolong sample storage glutamine and asparagine decrease whereas glutamic and aspartic acids increase simultaneously [3]. Other factors such as urinary bacterial contamination can significantly alter urinary amino acids profile [23]. Regarding all this, a complete interpretation of an amino acid profile should be made in light of the patient’s clinical and medical history.
Conclusions
In our study, over 27% of patients showed abnormalities in amino acid profile. The diagnosis of Phenylketonuria and Non-ketotic hyperglycinemia has been established at the biochemical and genetic level. Elevated concentration of Ala represents a biomarker for a metabolic pathology with mitochondrial involved. Based on performed metabolic workup, all reported patients are on treatment with positive outcome. The early and precise diagnosis of IEM is essential for the long-term health of affected subjects.
Competing interests
None declared
Authors’ contribution
VH, CB elaborated the hypothesis and design of the study. NU, DB gathered primary material, selected the patients according to inclusion and exclusion criteria. EE, CD and AN had a significant contribution in the analysis performing and interpretation of the data. NU revised the manuscript critically. All authors have read and approved the final version of the manuscript.
Acknowledgment
Isabela Tarcomnicu, Danae Stambouli from CytoGenomic Medical Laboratory, Bucharest, Romania for performing metabolic extended screening from dried blood spots by liquid chromatography-tandem mass spectrometry (LC-MS/MS);
Svetlana Garaeva, Galina Postolati from Institute of Physiology and Sanocreatology of Academy of Science of Moldova, Chisinau, Moldova for performing ion-exchange liquid chromatography.
Authors’ ORCID IDs
Victoria Hlistun - https://orcid.org/0000-0002-8563-3418
Egor Efremov - https://orcid.org/0000-0002-4544-353X
Daniela Blaniță - https://orcid.org/0000-0001-7736-3406
Chiril Boiciuc - https://orcid.org/0000-0002-7273-2492
Calin Deleanu - https://orcid.org/0000-0001-8206-5227
Alina Nicolescu - https://orcid.org/0000-0001-7022-8893
Natalia Usurelu - https://orcid.org/0000-0001-8685-393
References
Ușurelu N. Era Erorilor Înnăscute de Metabolism în Moldova, Bul. Perinatol., 2016; 1(69), 54–58.
Ferreira C. R., Van Karnebeek C. D., Vockley J., Blau N., A proposed nosology of inborn errors of metabolism. Genet Med., 2019; 1(21), 102–106.
Sandlers Y., Amino Acids Profiling for the Diagnosis of Metabolic Disorders. Biochemical Testing-Clinical Correlation and Diagnosis, 2019.
Wasim M., Awan F. R., Khan H. N., Tawab A., Iqbal M., Ayesha H., Aminoacidopathies: Prevalence, Etiology, Screening, and Treatment Options. Biochem. Genet., 2018; 1(56), 7–21.
Scaturro G., Sanfilippo C., Piccione C., Piro E, Giuffrè M, Corsello G. Newborn screening of inherited metabolic disorders by tandem mass spectrometry: past, present and future. Pediatr Med Chir., 2013; 35(3), 105-109.
Waters D., Adeloye D., Woolham D., Wastnedge E., Patel S., Rudan I., Global birth prevalence and mortality from inborn errors of metabolism: A systematic analysis of the evidence. J. Glob. Health, 2018; 2(8).
Wertheim-Tysarowska K., Gos M., Sykut-Cegielska J., Bal J. Genetic analysis in inherited metabolic disorders--from diagnosis to treatment. Own experience, current state of knowledge and perspectives. Dev. period Med., 2015; 4(19), 413–431.
Ezgu F. Inborn Errors of Metabolism. Adv Clin Chem,. 2016; 1(73), 195-250.
Ușurelu N., Introduction into Inborn Errors of Metabolism. Bul. Perinatol., 2020; 1(86), 7–11.
Vernon H. J., Inborn errors of metabolism: Advances in diagnosis and therapy. JAMA Pediatr., 2015; 8(169), 778–782.
Ismail I. T., Showalter M. R., Fiehn O. Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites, 2019; 9(242), 1–25.
Zschocke J., Hoffman G. F., Vademecum Metabolicum, 3rd Edition. Milupa Metab., 2011; p. 175.
Krieger I., Winbaum E. S., Eisenbrey A. B. Cerebrospinal fluid glycine in nonketotic hyperglycinemia. Effect of treatment with sodium benzoate and a ventricular shunt. Metabolism, 1977; 5(26), 517–524.
Swanson M. A., Coughlin C. R. et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann. Neurol., 2015; 4(78), 606–618.
Ușurelu N., Țurea V, Ușurelu O, Sacară V., Gavriliuc A. Fenilcetonuria:consultul medico-genetic, dietoterapia, integrarea socială (ghid practic), 2008; 33-51.
Van Wegberg A. M. J. et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis., 2017; 1(12), 1–56.
Sharman R., Sullivan K., Young R., Mcgill J. A preliminary investigation of the role of the Phenylalynine:Tyrosine ratio in children with early and continuously treated Phenylketonuria: Toward identification of ‘Safe’ levels. Dev. Neuropsychol., 2010; 1(35), 57–65.
Levy P. A. Inborn errors of metabolism: Part 1: Overview. Pediatr. Rev., 2009; 4(30), 131–138.
Tansek M. Z., Groselj U., Angelkova N., Anton D., Baric I., Djordjevic M.. Phenylketonuria screening and management in southeastern Europe – survey results from 11 countries. Orphanet J Rare Dis. 2015; 10(1), 1–7.
Eastman J. W., Sherwin J. E., Wong R., Liao C. L., Currier R. J., Lorey F., Cunningham G. Use of the phenylalanine:tyrosine ratio to test newborns for phenylketonuria in a large public health screening programme. J Med Screen. 2000; 7(3), 131-135.
Garg U., Smith L. D. Biomarkers in Inborn Errors of Metabolism. 2017.
Milsom J. P., Morgan M. Y., Sherlock S. Factors affecting plasma amino acid concentrations in control subjects. Metabolism, 1979; 4(28), 313–319.
Vidler J., Wilcken B. Prevalence of unsuspected urinary bacterial contamination: Effects of screening tests for detection of inborn errors of metabolism. Clin. Chim. Acta, 1978; 1(82), 173–178, 1978.